
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An expanded MIL-53-type coordination polymer with a reactive pendant ligand†
Hannah
Kunicki
a,
Thomas W.
Chamberlain
a,
Guy J.
Clarkson
a,
Reza J.
Kashtiban
b,
Joseph E.
Hooper
c,
Daniel M.
Dawson
c,
Sharon E.
Ashbrook
 c and
Richard I.
Walton
*a
c and
Richard I.
Walton
*a
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: r .i.walton@warwick.ac.uk
bDepartment of Physics, University of Warwick, Coventry, CV4 7AL, UK
cSchool of Chemistry, and EaStCHEM University of St Andrews, North Haugh, St Andrews, KY16 9ST, UK
First published on 18th July 2018
Abstract
A coordination polymer with a structure isoreticular to MIL-53, but containing a divalent metal cation (Co2+ or Mg2+) and a coordinating 4-styrylpyridine-N-oxide ligand, crystallises under solvothermal conditions; the pendant ligand can be brominated, with evidence from powder X-ray diffraction, element mapping and 81Br solid-state NMR for this post-synthesis modification.
Materials with the MIL-53 structure represent one of the most widely studied of all families of metal–organic frameworks. The simple structure, containing infinitely linked trans-corner shared octahedral metal centres cross-linked by bridging 1,4-benzene-dicarboxylate linkers, has an open net with diamond-shaped channels running parallel to the metal oxide chains.1 This ‘wine-rack’ structure is now renowned for its structural flexibility, where, in response to pressure, temperature or molecular guests, variation in the hinge angles of the link of the ligand to the metals allows a reversible opening and closing of the structure.2 This so-called ‘breathing’ effect has been widely investigated for the capture and release of small molecules in the gas and liquid phase, offering responsive, porous adsorbents,3 a property now realised and rationalised using computer simulation.4 The structure was first reported in a vanadium form, also known as MIL-47, where oxidation leads to tetravalent metal cations bridged by oxide anions in the inorganic chain,5 but most work has been done on MIL-53 materials containing trivalent cations (such as Cr3+,6 Al3+,7 Fe3+,8 and Ga3+ (ref. 9)), with hydroxide as the bridging anion. A related set of materials can be formed for divalent cations (Co2+, Mn2+, Mg2+) where the atom bridging the metals in the inorganic chain is the oxygen of a pyridine-N-oxide:10 this pendant ligand may occupy the channel space of the framework, rendering the structure non-porous, but its steric bulk can be used to distort the framework, further illustrating the inherent flexibility of the MIL-53 structure.11
An isoreticular form of MIL-53, in which the 1,4-benzene-dicarboxylate linker is replaced by the extended 4,4′-biphenyl dicarboxylate linker was reported by Senkovska et al. and named DUT-5.12 Although the parent aluminium form of the material was reported to have permanent porosity with a rigid, non-breathing structure, a vanadium form COMOC-2, showed pore opening and closing with gas pressure,13 suitable for separation of hydrocarbons.14 In this communication we describe how an expanded form of MIL-53 can be generated from divalent metals and 4,4′-biphenyl dicarboxylate in combination with an extremely bulky pendant pyridine-N-oxide ligand, which in turn provides the possibility of introducing reactive functionality to the framework for post-synthesis chemistry.
The ligand 4-styrylpyridine-N-oxide (4S-PNO) was synthesised via 4-styrylpyridine using a method adapted from the literature (see ESI†). Initial attempts to crystallise a coordination polymer with the 4S-PNO ligand and 1,4-benzene-dicarboxylate, as in previous work on divalent metals and pyridine-N-oxides,10 yielded only a co-crystal of the ligand and 1,4-benzene-dicarboxylic acid (ESI†), with the metal ions presumably remaining in solution, so we instead selected the extended 1,4-biphenyl dicarboxylate (BPDC) linker. Thus for either Co2+ or Mg2+ isostructural coordination polymers M(4S-PNO)(BPDC)·nH2O were isolated as single crystals suitable for structure determination (ESI†). The amount of water in the magnesium form was found to be negligible, but extra-framework electron density in the cobalt form could be accounted for by 0.25 water molecules per formula unit. Fig. 1a shows a view of the crystal structure of Co(4S-PNO)(BPDC)·0.25H2O illustrating the 1-dimensional channels, with Fig. 1b and c perpendicular views, showing the connectivity of the structure and the orientation of the pendant 4S-PNO ligand.
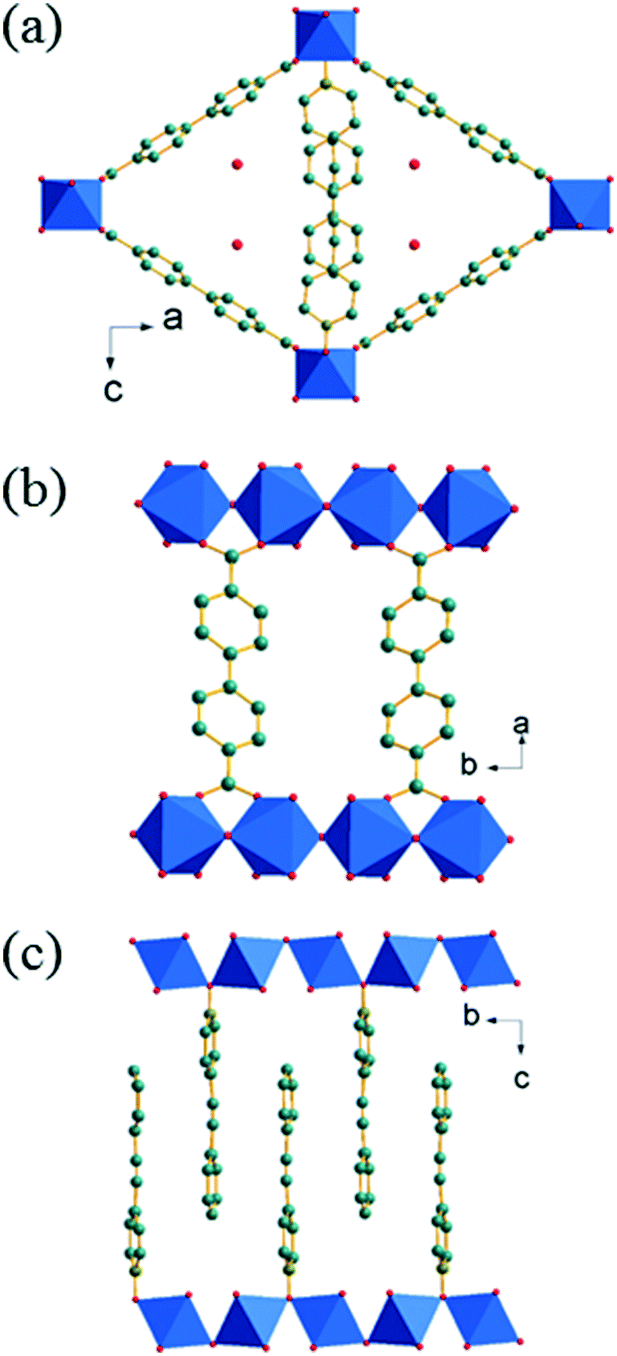 | ||
| Fig. 1 Structure of M(4S-PNO)(BPDC) (M = Mg, Co) drawn using the coordinates of the Co form of the material: (a) parallel to the inorganic chains, and (b) and (c) in perpendicular directions, showing the orientation of the BPDC linker and 4S-PNO ligand, respectively. The metal-centred octahedra are shaded blue with carbon, nitrogen and oxygen as grey, olive and red spheres, respectively. Only one orientation of the disordered 4S-PNO and BPDC are shown in each case and the partially filled water sites are shown in (a). | ||
Fig. 2 compares the structures of the Mg and Co forms of the new coordination polymer with the related materials DUT-5 and COMOC-2, with structural features identified to provide a quantitative comparison (Table 1). This shows that while COMOC-2 presents a fully open structure (D1/D2 ≈ 1), and DUT-5 is close to being fully open, the new materials have partially open frameworks. There is no evidence for intermolecular interactions between the pendant 4S-PNO ligands, such as π–π stacking, since the aryl groups between neighbouring ligands are not aligned and are separated by ∼4 Å. The degree of opening of the structure apparently depends both on the hinge angle between the carboxylate and the pairs of metals that it bridges (α) and also the intra-chain M–O–M angle (β), with the most open forms of the framework having almost completely open ‘hinges’ (α ≈ 180°) and larger M–O–M angles. Without further examples of materials with these structures, it is difficult to draw more detailed conclusions about any correlations between these structural parameters.
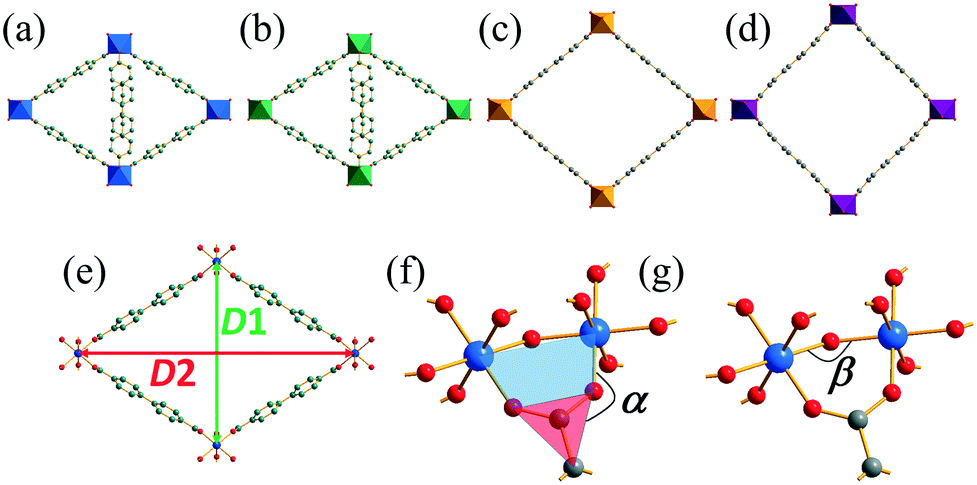 | ||
| Fig. 2 Comparison of the structures of (a) Co(4S-PNO)(BPDC)·0.25H2O (b) Mg(4S-PNO)(BPDC) (c) DUT-5 and (d) COMOC-2, with relevant interatomic distances and angles defined in (e)–(g). | ||
The phase purity of M(4S-PNO)(BPDC) (M = Mg, Co) was confirmed by powder X-ray diffraction (XRD), while thermogravimetric analysis corroborated the identity of the bulk sample (for M = Mg, expected total mass loss on combustion to MgO is 91.3%, observed 89.7%, while for M = Co, expected total mass loss on combustion to CoO is 85.1%, observed 88.6%). Thermodiffractometry in flowing air was then used to investigate the phase stability and the possibility of any thermally induced structural deformation, as shown in Fig. 3. This shows how the material collapses just above 200 °C, and despite continued evolution of the mass loss curve seen by thermogravimetric analysis (TGA) until above 500 °C there is complete loss of crystallinity by 250 °C. Prior to collapse there are no shifts in the Bragg peak positions indicating that the framework is rigid and non-breathing, i.e. , expansion to give a fully open framework, as in COMOC-2, does not occur with temperature.
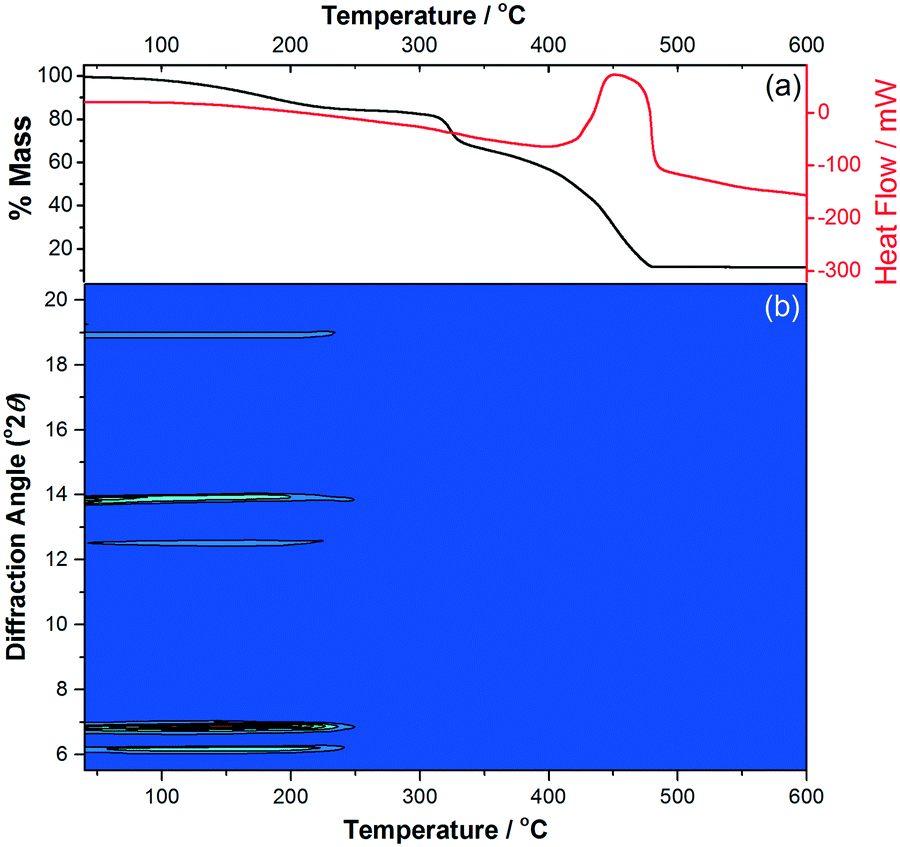 | ||
| Fig. 3 (a) TGA-DSC and (b) X-ray thermodiffractometry (Cu Kα) in flowing air of Co(4S-PNO)(BPDC)·0.25 H2O showing the absence of framework expansion before collapse. | ||
Previous modifications of the DUT-5 structure have involved adding functionality to the BPDC linker: for example, sulfone modified versions were prepared for tuning the adsorption of hydrocarbons,15 while a set of functionalised linkers variants were prepared and post synthesis modifications performed.16 For our new material, the pendant ligand has the potential as a site for a distinctly different type of post-synthesis modification. We thus investigated the bromination of this group to establish this possibility. The magnesium form of the material was chosen since it is white, allowing for any effect of bromination to be visually verified, and diamagnetic, allowing spectroscopic observation using solid-state NMR. Furthermore, the presence of a redox inactive cation would minimise additional side reactions occurring. A sample of 150 mg Mg(4S-PNO)(BPDC) was stirred in a solution of bromine (100 mg) in chloroform (5 mL) for 3 days in darkness. The recovered solid, yellow in colour, was allowed to dry in air at room temperature before characterisation. Powder XRD clearly shows that the framework of the material is intact after bromine treatment, with little shift in Bragg peak positions, Fig. 4a, but that the relative intensity of the Bragg peaks is dramatically altered. In particular, the low-angle peaks noticeably reduced in intensity compared to those at higher angle, and this is entirely consistent with the presence of higher electron density species within the structure, as seen for zeolites, for example, when extra-framework space is occupied.17 The powder XRD does show some loss of crystallinity, however, and the presence of an amorphous background, which limits quantifying structural information. Further evidence for the inclusion of bromine is provided by element mapping using energy dispersive X-ray analysis on the scanning electron microscope, Fig. 4b and ESI.† This shows that the bromine is dispersed throughout the crystallites analysed as homogeneously as the other elements present. Bulk chemical analysis shows the presence of 15.8% Br by mass, which would correspond to, on average, just over one half of the carbon–carbon double bonds being dibrominated to give chemical formula Mg(C13H10BrNO)(BPDC) with an expected Br content of 14.8%. Solution-state NMR spectroscopy of an acid-digested sample, a method that has been commonly used to analyse the post-synthetic modification of MOFs, including in bromination studies,18 was inconclusive for our material. This may in part be due to the reactivity of the 4S-PNO ligand and its complex fragmentation in acid solution, and indeed this is a shortcoming of the acid-digestion method for analysis of reactive organic components. As an alternative, we investigated the use of 13C and 81Br solid-state NMR spectroscopy to provide evidence of the chemical nature of the bromine in the modified MOF. 13C MAS NMR spectra indicated retention of the organic components of the framework and a structural change, although no direct evidence of C–Br bonds (see ESI†). 81Br is not widely studied due to its large quadrupole moment and Sternheimer antishielding factor, yielding extremely broad spectral resonances,19 and using high field (20.0 T) wideline WURST-QCPMG experiments20 our measured spectrum reveals a very broad signal, Fig. 4c, that is not inconsistent with the presence of C–Br bonds. Importantly there is no evidence for the presence of MgBr2,21 which may have conceivably formed upon breakdown of the MOF in contact with bromine.
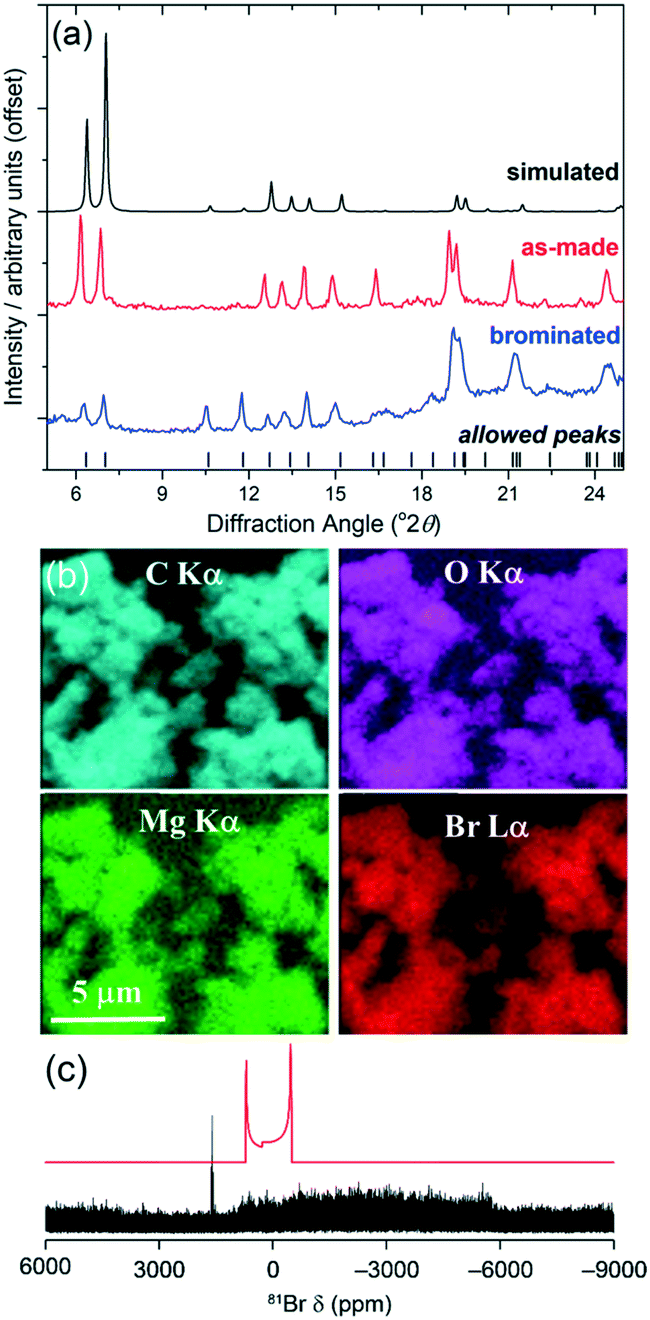 | ||
| Fig. 4 Characterisation of brominated Mg(4S-PNO)(BPDC): (a) powder XRD, (b) EDXA mapping, and (c) 81Br wideline solid-state NMR with the offset simulated spectrum of MgBr2 in red. | ||
Despite the bulk of the pendant ligand calculation of the potential void space in the structure (see ESI†) shows that ∼25.0% of the unit cell volume is free space, and, furthermore, when considering the one-dimensional nature of the channels in which the ligand resides, this could be accessible (see ESI† for a space-filling representation of the structure). This would explain how bromination of the ligand is possible.
The literature now contains various reports of post-synthetic bromination of MOFs including by reaction between bromine and carbon–carbon double or triple bonds.18 All of these, however, are associated with the linker making up the MOF structure and make use of highly porous frameworks. For example, Wang and Cohen used a 2-amino-1,4-benzenedicarboxylate as linker to prepare IRMOF-5 and then introduced bromine via an alkenyl linker modification in a tandem process,18a while Marshall et al. prepared an isoreticular version of zirconium UiO-66 with a 4,4-ethynylenedibenzoate linker that could be dibrominated to modify the mechanical properties of the framework.18e,f For the material described herein, the use of a pendant co-ligand provides a distinct approach for the introduction of a reactive site in metal carboxylate frameworks for post-synthesis modification. This will be of relevance to the design of new materials that may have applications in sensing, catalysis or molecular capture. It would also be interesting to investigate the preparation of analogous materials using other cations, or mixtures of cations, since in the case of MIL-53 this has been proven to have an influence on the structural flexibility of the framework and so may allow the properties to be tuned.22
We are grateful to the University of Warwick Undergraduate Research Support Scheme for award of a bursary to TWC. The EPSRC's UK National Crystallography Service at the University of Southampton is thanked for one of the data sets. The UK 850 MHz solid-state NMR Facility was funded by EPSRC and BBSRC (contract reference PR140003), as well as the University of Warwick including via part funding through Birmingham Science City Advanced Materials Projects 1 and 2 supported by Advantage West Midlands (AWM) and the European Regional Development Fund (ERDF); collaborative assistance from the Facility Manager Dinu Iuga is acknowledged. SEA would like to thank the ERC (EU FP7 Consolidator Grant 614290 “EXONMR”). The research data supporting this publication are available at: http://wrap.warwick.ac.uk/103572
Conflicts of interest
There are no conflicts to declare.Notes and references
- F. Millange, C. Serre and G. Férey, Chem. Commun., 2002, 822 RSC.
- G. Férey and C. Serre, Chem. Soc. Rev., 2009, 38, 1380 RSC.
- (a) S. Bourrelly, P. L. Llewellyn, C. Serre, F. Millange, T. Loiseau and G. Férey, J. Am. Chem. Soc., 2005, 127, 13519 CrossRef PubMed; (b) F. Millange, C. Serre, N. Guillou, G. Férey and R. I. Walton, Angew. Chem., Int. Ed., 2008, 47, 4100 CrossRef PubMed.
- F. Salles, A. Ghoufi, G. Maurin, R. G. Bell, C. Mellot-Draznieks and G. Férey, Angew. Chem., Int. Ed., 2008, 47, 8487 CrossRef PubMed.
- K. Barthelet, J. Marrot, D. Riou and G. Férey, Angew. Chem., Int. Ed., 2002, 41, 281 CrossRef PubMed.
- C. Serre, F. Millange, C. Thouvenot, M. Nogues, G. Marsolier, D. Louer and G. Férey, J. Am. Chem. Soc., 2002, 124, 13519 CrossRef PubMed.
- T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Férey, Chem. – Eur. J., 2004, 10, 1373 CrossRef PubMed.
- (a) T. R. Whitfield, X. Q. Wang, L. M. Liu and A. J. Jacobson, Solid State Sci., 2005, 7, 1096 CrossRef; (b) F. Millange, N. Guillou, R. I. Walton, J. M. Grenèche, I. Margiolaki and G. Férey, Chem. Commun., 2008, 4732 RSC.
- C. Volkringer, T. Loiseau, N. Guillou, G. Férey, E. Elkaim and A. Vimont, Dalton Trans., 2009, 2241 RSC.
- (a) G. H. Xu, X. G. Zhang, P. Guo, C. L. Pan, H. J. Zhang and C. Wang, J. Am. Chem. Soc., 2010, 132, 3656 CrossRef PubMed; (b) A. S. Munn, S. Amabilino, T. W. Stevens, L. M. Daniels, G. J. Clarkson, F. Millange, M. J. Lennox, T. Duren, S. Bourelly, P. L. Llewellyn and R. I. Walton, Cryst. Growth Des., 2015, 15, 891 CrossRef; (c) A. S. Munn, G. J. Clarkson, F. Millange, Y. Dumont and R. I. Walton, CrystEngComm, 2013, 15, 9679 RSC; (d) Y. X. Liu, D. Liu and C. Wang, J. Solid State Chem., 2015, 223, 84 CrossRef.
- A. S. Munn, G. J. Clarkson and R. I. Walton, Acta Crystallogr.ect. B: Struct. Sci.ryst. Eng. Mater., 2014, 70, 11 CrossRef PubMed.
- I. Senkovska, F. Hoffmann, M. Froba, J. Getzschmann, W. Bohlmann and S. Kaskel, Microporous Mesoporous Mater., 2009, 122, 93 CrossRef.
- Y. Y. Liu, S. Couck, M. Vandichel, M. Grzywa, K. Leus, S. Biswas, D. Vollmer, J. Gascon, F. Kapteijn, J. F. M. Denayer, M. Waroquier, V. Van Speybroeck and P. Van der Voort, Inorg. Chem., 2013, 52, 113 CrossRef PubMed.
- S. Couck, T. R. C. Van Assche, Y. Y. Liu, G. V. Baron, P. Van der Voort and J. F. M. Denayer, Langmuir, 2015, 31, 5063 CrossRef PubMed.
- (a) S. Couck, Y. Y. Liu, K. Leus, G. V. Baron, P. Van der Voort and J. P. M. Denayer, Microporous Mesoporous Mater., 2015, 206, 217 CrossRef; (b) G. Wang, K. Leus, S. Couck, P. Tack, H. Depauw, Y. Y. Liu, L. Vincze, J. F. M. Denayer and P. Van der Voort, Dalton Trans., 2016, 45, 9485 RSC.
- M. A. Gotthardt, S. Grosjean, T. S. Brunner, J. Kotzel, A. M. Ganzler, S. Wolf, S. Brase and W. Kleist, Dalton Trans., 2015, 44, 16802 RSC.
- R. S. P. King, P. F. Kelly, S. E. Dann and R. J. Mortimer, Chem. Commun., 2007, 4812 RSC.
- (a) Z. Q. Wang and S. M. Cohen, Angew. Chem., Int. Ed., 2008, 47, 4699 CrossRef PubMed; (b) S. C. Jones and C. A. Bauer, J. Am. Chem. Soc., 2009, 131, 12516 CrossRef PubMed; (c) F. Sun, Z. Yin, Q. Q. Wang, D. Sun, M. H. Zeng and M. Kurmoo, Angew. Chem., Int. Ed., 2013, 52, 4538 CrossRef PubMed; (d) D. Frahm, F. Hoffmann and M. Froba, Cryst. Growth Des., 2014, 14, 1719 CrossRef; (e) R. J. Marshall, S. L. Griffin, C. Wilson and R. S. Forgan, J. Am. Chem. Soc., 2015, 137, 9527 CrossRef PubMed; (f) R. J. Marshall, T. Richards, C. L. Hobday, C. F. Murphie, C. Wilson, S. A. Moggach, T. D. Bennett and R. S. Forgan, Dalton Trans., 2016, 45, 4132 RSC; (g) X. Y. Xu, F. Yang, S. L. Chen, J. Y. He, Y. Q. Xu and W. Wei, Chem. Commun., 2017, 53, 3220 RSC.
- D. L. Bryce, C. M. Widdifield, R. P. Chapman and R. J. Attrell, in NMR of Quadrupolar Nuclei in Solid Materials, ed. R. E. Wasylishen, S. E. Ashbrook and S. Wimperis, John Wiley and Sons, ChichesterK, 2012 Search PubMed.
- L. A. O'Dell and R. W. Schurko, Chem. Phys. Lett., 2008, 464, 97 CrossRef.
- C. M. Widdifield and D. L. Bryce, Phys. Chem. Chem. Phys., 2009, 11, 7120 RSC.
- F. Nouar, T. Devic, H. Chevreau, N. Guillou, E. Gibson, G. Clet, M. Daturi, A. Vimont, J. M. Grenèche, M. I. Breeze, R. I. Walton, P. L. Llewellyn and C. Serre, Chem. Commun., 2012, 48, 10237 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Ligand synthesis, details of crystal structures and further characterisation data. The crystal structures are deposited as CCDC 1842156–1842158. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ce00891d |
| This journal is © The Royal Society of Chemistry 2018 |