
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of phosphorylation and dephosphorylation of shell matrix proteins in shell formation: an in vivo and in vitro study†
Jinzhe
Du‡
a,
Guangrui
Xu‡
ac,
Chuang
Liu
 *bc and
Rongqing
Zhang
*ac
*bc and
Rongqing
Zhang
*ac
aInstitute of Marine Biotechnology, School of Life Sciences, Tsinghua University, Beijing 100084, China
bDepartment of Biomaterials, Max Planck Institute of Colloids and Interfaces, Potsdam 14476, Germany. E-mail: Chuang.Liu@mpikg.mpg.de; rqzhanglab@mail.tsinghua.edu.cn; Tel: +86 10 62772630
cDepartment of Biotechnology and Biomedicine, Yangtze Delta Region Institute of Tsinghua University, Jiaxing, Zhejiang Province 314006, China
First published on 1st June 2018
Abstract
Protein phosphorylation is a fundamental mechanism regulating many aspects of cellular processes. Shell matrix proteins (SMPs) control the crystal nucleation, polymorphism, morphology, and organization of calcium carbonate crystallites during shell formation. SMP phosphorylation is suggested to be important in shell formation but the mechanism is largely unknown. Here, to investigate the mechanism of phosphorylation of SMPs in biomineralization, we performed in vivo and in vitro experiments. By injection of an antibody against the anti-phosphoserine/threonine /tyrosine sites into the extrapallial fluid of the pearl oyster Pinctada fucata, phosphorylation of matrix proteins was significantly reduced after 6 days. Newly formed prismatic layers and nacre tablets were found to grow abnormally with reduced crystallinity and possibly changed crystal orientation shown by Raman spectroscopy. In addition, regeneration of shells is also inhibited in vivo. Then, protein phosphatase was used to dephosphorylate SMPs extracted from the shells. After dephosphorylation, the ability of SMPs to inhibit calcium carbonate formation has been reduced. Surprisingly, the ability of SMPs to modulate the crystal morphology has been largely compromised although the phosphorylation extent remained as at least half that of the control. Furthermore, dephosphorylation of SMPs changed the distribution of protein occlusions and decreased the amount of protein occlusions inside the crystals shown by confocal imaging, indicating the interaction between phosphorylated SMPs and crystals. Taken together, this study provides insight into the mechanism of phosphorylation of SMPs during shell formation.
Introduction
Biomineralization is the process by which living organisms produce minerals, and includes the formation of silicates in algae and diatoms, carbonates in invertebrates, and calcium phosphates and carbonates in the hard tissues of vertebrates.1 Macromolecules have been reported to play central roles in the formation of various biominerals.2–5 Shells, made of calcium carbonate, are typical examples of biominerals. For example, nacre possesses superior mechanical and biological properties compared to its counterpart minerals.6 Shell matrix proteins (SMPs), extracted from shells, play important roles in the crystal nucleation, polymorphism, morphology, and organization of calcium carbonate crystallites during shell formation.7,8 Previous studies have been focused on studying the role of SMPs expressed in host cells such as Escherichia coli in mineralization while the role of protein post-translational modifications (PTMs) in shell formation is still lacking.9–13 For example, previously, our group demonstrated that PfN23 increased the rate of calcium carbonate deposition and induced the formation of aragonite crystals.9 Bahn So Yeong et al. showed that Pif80 participated in the formation of polymer-induced liquid precursor-like amorphous calcium carbonate granules and stabilized these granules.14It is noted that some SMPs have PTMs such as glycosylation, phosphorylation and sulfation, which are crucial for their functions.15 Among them, phosphorylation is one of the most widespread protein PTMs and also occurs in the organic matrix of biominerals.16,17 Previously, Fam20C was found to be a serine protein kinase located in Golgi and was able to phosphorylate proteins that regulate biomineralization.18 Interestingly, osteopontin (OPN), a secreted, glycosylated phosphoprotein found in mineralized tissues such as bones and teeth,19 has been intensively studied. It is found that phosphorylation of OPN is required for inhibition of vascular smooth muscle cell calcification. Moreover, phosphorylated OPN peptides can inhibit mineralization of calcium phosphate20 and calcium oxalate.21 OPN is also a major component of the calcium carbonate-containing egg shell, which inhibited calcite growth and produced altered crystal morphology in vitro.22
Some studies attempted to probe the role of protein phosphorylation in invertebrate biomineralization. Borbas et al. showed that a phosphorylated shell matrix was more effective in inhibiting crystal growth than a dephosphorylated matrix.23 Interestingly, one third of the Ser/Thr phosphorylated sites with family with sequence similarity 20C (Fam20C) recognition site Ser-x-Glu were found in the Lottia gigantea shell matrix proteome,24 indicating their potential function in biomineralization. However, the detailed mechanism for the effect of phosphorylation on shell formation or calcium carbonate formation is limited. Previously, we have shown that knockdown of Fam20C, which can phosphorylate biomineralization related proteins in vivo, resulted in abnormal stacking of calcium carbonate crystals at the edges of nacre tablets.25 These results suggest that phosphorylation is important in shell formation.
Phosphorylation of proteins can affect their interaction with calcium and possibly their occlusion inside the crystals. Occlusion of proteins or organics have been observed in many biominerals including nacre,26–28 sea urchin spines29 and coccoliths.4 The occlusion of proteins or organics is related to their remarkable mechanical properties.30 However, the relationship between protein phosphorylation and protein occlusion is still unknown. Therefore, in this study, we aim to investigate the role of SMP phosphorylation during shell formation by using both in vivo and in vitro experiments.
Materials and methods
Sample preparation
All pearl oysters Pinctada fucata used in this study were collected from Zhanjiang, Guangdong province of China, and were cultured at 20 degrees centigrade in artificial seawater (3% salinity).Injection of antibody into the extrapallial fluid of P. fucata
2 μg of anti-phosphoserine/threonine/tyrosine antibody (Beijing Biosynthesis Biotechnology Co., LTD.) were diluted in 200 μL PBS. The diluted antibody was injected into the extrapallial fluid of ten oysters as the experimental groups. At the same time, diluted anti-BSA (Abcam) was injected into the extrapallial fluid of ten oysters as the control group. PBS was also injected into the extrapallial fluid of ten oysters as a negative control. Injection was carried out by using a 0.8 × 50 mm syringe needle through the byssal foramen per day for 6 days. The extrapallial fluid of five oysters was extracted after 3 days and the extrapallial fluid of another five oysters was extracted after 6 days to examine the phosphorylation level of matrix proteins. Then, the oysters were executed. The inner surface of the newly formed shells of oysters after injection for 3 days and 6 days was naturally dried and sampled to be observed by SEM. Representative pictures were chosen to be presented.Shell notching experiment
The shell notching of pearl oysters was performed as described by Mount et al.31 Five anti-phosphoserine/threonine/tyrosine injected oysters, five anti-BSA injected oysters and five PBS injected oysters were notched without injuring the mantle tissues. Injection was carried out per day for 6 days. Notching was performed immediately following the first day injection. The extrapallial fluid of half of the oysters was extracted after 3 days and the extrapallial fluid of the other half of the oysters was extracted after 6 days to examine the phosphorylation level of matrix proteins. The regenerated shells were observed after notching for 6 days.Raman spectroscopy
The Raman spectra were recorded at an excitation wavelength of 532 nm. The spectra were scanned three times for 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) s in the range of 100–1400cm−1 using a LabRAM HR Evolution spectrometer (HORIBA Jobin Yvon).
s in the range of 100–1400cm−1 using a LabRAM HR Evolution spectrometer (HORIBA Jobin Yvon).
Extraction of SMPs from the shells of P. fucata
The procedure is according to a previously established protocol in our group.32 Briefly, the cleaned shells of P. fucata were immersed in a 5% NaOH solution for 24h followed by rinsing with distilled water to avoid possible contamination from soft tissues. The inner aragonite nacreous layer was separated mechanically by abrasion after air-drying. The fragments were pulverized (30g) and then were decalcified using 0.8M EDTA (pH = 8.0) at 4 °C for 60h under continuous stirring. For the extraction of the EDTA soluble matrix (ESM) of the nacreous layer, the supernatant was collected by centrifugation at 13000rpm at 4 °C for 30min and then desalted by ultrafiltration (3kDa, 4000rpm, 1–2h). For the extraction of the insoluble matrix (EISM), the above precipitates were thoroughly rinsed with water and were incubated with denaturing solution (30 mM Tris-HCl, pH 8.0, 1% SDS, 10 mM dithiothreitol) at 100 °C for 30 min. The concentration of proteins was determined using a Nanodrop 2000 (ThermoFisher Scientific, USA) or the bicinchoninic acid assay method.
Dephosphorylation of SMPs by lambda protein phosphatase (PPase)
Extracted proteins were incubated with PPase (New England Biolabs, USA) following instructions. 20 μg of proteins were incubated with 400 units PPase in a 50 μL system at 30 °C for 30 min. 5 μL phosphatase buffer, 5 μL 1 mM MnCl2 and 0.5 μL protease inhibitor were also added. After reaction, dephosphorylated proteins were dialyzed overnight using a mini D-tube (Novagen, cutoff 3 K) to remove the buffer.Detection of phosphorylation by enzyme linked immunosorbent assay (ELISA)
Corning high affinity EIA/RIA 96 well plates (Corning Inc., NY, USA, #9018) were coated with 500 ng SMPs or dephosphorylated SMPs per well at 4 °C overnight. Wells coated with PBS were used as a negative control. After blocking with 5% BSA in PBS at 25 °C for 2 h, 500 times diluted rabbit anti-phosphoserine/threonine/tyrosine (Beijing Biosynthesis Biotechnology Co., LTD.) used as the primary antibody was incubated on the plate for 2 h at 25 °C. HRP-coupled goat anti-rabbit-IgG (Sino Biological Inc.) was used as the secondary antibody and was incubated for 2 h at 25 °C before addition of the 3,3′,5,5′-tetramethylbenzidine (TMB) super sensitive one component HRP microwell substrate (Surmodics, USA). 2 M H2SO4 was used to terminate the reaction. The plates were analyzed with a spectrophotometer (Bio-Rad 680) at 450 nm. The wells were washed five times with ELISA-washing buffer (PBS with 0.05% Tween 20) between each step of the ELISA. All antibodies were diluted in ELISA incubation buffer (PBS with 0.05% Tween 20, 0.5% BSA).Inhibition of calcium carbonate precipitation assay
The effect of dephosphorylated SMPs on the rate of calcium carbonate precipitation was tested according to the method of Suzuki et al.33 with some modifications. The protein (the final concentration was 10 μg mL−1) was mixed with 100 μL of 100 mM sodium bicarbonate, pH 8.5. After the addition of 100 μL of 100 mM calcium chloride to the mixed solution, the formation of calcium carbonate precipitate was monitored by recording the changes in the turbidity every 30 seconds for 5 min based on the absorbance at 570 nm measured using a spectrophotometer (Bio-Rad 680).Labeling ESM proteins by Cy5
The procedure is according to a previous protocol.32N-Hydroxysuccinimide (NHS) esters and other activated esters (sulfo-NHS; sulfotetrafluorophenyl, STP) are reactive compounds suitable for the modification of amino groups. To label proteins with Cy5, 0.14 mg of NHS-Cy5 (LiTTLE-PA Sciences Company, China) was first dissolved in 100 μL of DMSO. Proteins were dissolved in 0.1 M sodium bicarbonate buffer (pH = 8.2). One hundred microliters of NHS-Cy5 ester was added into 900 μL of the 1.0 mg mL−1 protein solution, and the mixed solution was vortexed well, followed by incubation at room temperature for 4 h. To remove unreacted agents, the conjugates were dialyzed against water for 48 h at 4 °C under agitation in the dark. The resultant proteins were designated Cy5-ESM and Cy5-ESM-DP.In vitro calcite crystallization experiment by the ammonium carbonate method
In general, calcium carbonate was deposited onto cleaned cover glass or a glass bottom cell culture dish (Nest, HK) by the slow diffusion of approximately 2g of (NH4)2CO3 vapor in a closed desiccator for approximately 12h at room temperature. Each drop on the glass or the dish contained 180μL of 20mM CaCl2 and 20μL of proteins (200 μg mL−1). The proteins are ESM, dephosphorylated ESM, EISM and dephosphorylated EISM. DI water or BSA was also added as the control. The samples for further tests were rinsed twice with DI water and air-dried. In the literature, we found that the concentration of CaCl2 ranges from 7.5 mM to 25 mM, depending on different crystallization methods. In our previous study, we have showed that in the presence of 20 mM CaCl2 and in the absence of proteins, uniform crystals can be produced.
For confocal laser scanning imaging (CLSM), the Cy5-labeled ESM proteins (the final concentration was 10 μg mL−1) were added in the crystallization systems under the same experimental conditions.
Confocal laser scanning imaging (CLSM)
The CLSM images were obtained using an LSM 710 META confocal microscope (Zeiss, Germany), and the excitation wavelengths for Cy5-labeled samples were set at 647 nm. The gray value of fluorescence in crystals was quantified using software ImageJ.Scanning electron microscopy (SEM)
The morphologies of the samples were observed using a scanning electron microscope (SEM, JEOL JSM-6460LV, Japan) after sputter-coating with a thin layer of gold nanoparticles for 60 s.Phosphorylation site prediction
Serine, threonine or tyrosine phosphorylation sites were predicted using the NetPhos 3.1 Server (http://www.cbs.dtu.dk/services/NetPhos/). Nine EDTA-soluble matrix proteins (Nacrein, N16, Pif, Mantle gene 4, Heat shock protein 70, PNU7, PNU8, NU6 and MSI80) were tested.Statistical analysis
The T test method has been used for statistical analysis in this study using GraphPad Prism 6 software.Results
Phosphorylation of shell matrix proteins is critical for shell formation: in vivo experiment
First, phosphorylation sites based on protein sequences were predicted using the NetPhos 3.1 Server,34 with phosphorylation potential above 50% considered as valid. Nine typical SMPs (Nacrein, N16, Pif, Mantle gene 4, Heat shock protein 70, PNU7, PNU8, NU6 and MSI80) extracted from the shells were predicted (Fig. S1†). These proteins were identified in the shell proteome5 and some of them played critical roles in shell formation. For example, Pif, which was proved to regulate nacre tablet formation, was predicted to possess a large amount of phosphorylation sites.35 The results showed that these proteins all have phosphorylation sites especially in MSI80 and Nacrein (Fig. S1†).To examine the role of phosphorylation of SMPs during shell formation in vivo, we performed injection of an antibody against the anti-phosphoserine/threonine/tyrosine sites into the extrapallial fluid and observed the morphology of newly formed shells. The extrapallial fluid has been demonstrated to contain matrix proteins that play important roles in the formation of shells.36 Especially, it can facilitate the formation of a multilevel layered nacre-like structure on the pearl nucleus.37 The diagram of this experiment is shown in Fig. S2.† After injection, the phosphorylation of extrapallial fluid proteins by ELISA was detected, and the phosphorylation level of anti-phosphoserine/threonine/tyrosine injection oysters was reduced significantly by nearly 45% after 3 days and by 87% after 6 days (Fig. S3A†). The inner surface of the newly formed shells was observed by SEM. In the anti-phosphoserine/threonine/tyrosine injected group, compared with the control group (Fig. 1A–D), the morphology of the prismatic layer was not disturbed and showed a normal prism structure after antibody injection for 3 days (Fig. 1E and F), while after 6 days, the boundaries of prisms became unclear and some holes appeared within the prisms (Fig. 1G and H) and the growth of the prismatic layer was greatly disturbed. However, the morphology of the nacreous layer changed a lot when injecting anti-phosphoserine/threonine/tyrosine compared with the control group (Fig. 2A–D). After 3 days, overgrowth crystals accumulated randomly on the edge of the nacre tablets (Fig. 2E and arrowheads in 2F). And after 6 days, the number of random crystals became higher and even covered the whole nacre tablets (Fig. 2G and arrowheads in 2H). Some tablets became interconnected and lost normal hexagonal structures. We also measured the length of overgrowth crystals and calculated the ratio of the abnormal crystal to the whole nacre. It showed that the ratio of abnormal nacre increased significantly to about 20% after 3 days and 40% after 6 days (Fig. S4†).
 | ||
| Fig. 1 The morphology of the prismatic layer of P. fucata after antibody injection. SEM images of the newly formed inner surface of the prismatic layer in the anti-BSA injected group after 3 days (A and B) and 6 days (C and D). SEM images of the newly formed inner surface of the prismatic layer in the anti-phosphoserine/threonine/tyrosine injected group after 3 days (E and F) and 6 days (G and H). | ||
 | ||
| Fig. 2 The morphology of the nacreous layer of P. fucata after antibody injection. SEM images of the inner surface of the newly formed nacreous layer in the anti-BSA injected group after 3 days (A and B) and 6 days (C and D). SEM images of the inner surface of the newly formed nacreous layer in the anti-phosphoserine/threonine/tyrosine injected group after 3 days (E and F) and 6 days (G and H). The arrowheads in F and H refer to the overgrowth of nacre tablets. | ||
To confirm the polymorph of crystals in the newly formed prismatic layer and nacreous layer, we tested the abnormal prisms and abnormal nacre by Raman spectroscopy. The results showed no polymorph change in abnormal prisms and nacre compared with normal shells (Fig. 3A and C), but the Raman peaks at 157 cm−1 and 282 cm−1 declined in the abnormal prismatic layer (Fig. 3B), and at the same time, the Raman peaks at 144 cm−1, 190 cm−1, and 272 cm−1 declined in the abnormal nacreous layer (Fig. 3D). From the above, it is suggested that the degree of crystallinity in the abnormal prismatic layer and nacreous layer was decreased and the growth of newly formed shells became weakly regulated when the phosphorylation level of the extrapallial fluid proteins was reduced. The ratio of Raman peak intensity at 144 cm−1 and 154 cm−1 was also changed after the injection of antibodies (Fig. 3D), indicating a change of crystal orientation.38 However, this correlation is only empirical and as far as we know, the quantitative relationship between the orientation change and the intensity of Raman spectra is unknown. It is noted that this change is not consistent in different individual oysters as shown by different changes of Raman peak intensity (Fig. 3D).
 | ||
| Fig. 3 Raman spectroscopy of the newly formed shell of P. fucata. The inner surface of the newly formed prismatic layer (A) and nacreous layer (C) in the control and antibody groups after 6 days were tested by Raman spectroscopy. The zoom-in of A (100–350 cm−1) and C (100–400 cm−1) is shown in B and D, respectively. The red dotted boxes indicated the changed Raman peaks between the control and antibody groups. Antibody-1 and antibody-2 are two repeats in each group. The Raman intensity and peak ratio slightly varied between individuals. | ||
To examine the impact of phosphorylation on the ability of shell repairing, we performed shell notching together with antibody injection and detected the phosphorylation of extrapallial fluid proteins by ELISA, finding that the phosphorylation level of anti-phosphoserine/threonine/tyrosine injection oysters was significantly reduced by 5% after 3 days and by 85% after 6 days, respectively (Fig. S3B†). The regenerated shells of the anti-BSA injected group and anti-phosphoserine/threonine/tyrosine injected group were photographed. Out of five oysters in each group, four oysters regenerated shells in the control group (red arrows, Fig. 4A) and three oysters regenerated shells in the experimental group (red arrows, Fig. 4B). We calculated the pixel area of regenerated shells using the photographs and found that the area of regenerated shells in the experimental group was 40% smaller than that in the control group (Fig. 4C). The shell repair experiment showed that reduced phosphorylation of matrix proteins in the extrapallial fluid undermined the ability of shell repairing while the microstructure of newly formed shells (prismatic layers of the shells) is unaffected (Fig. S5†).
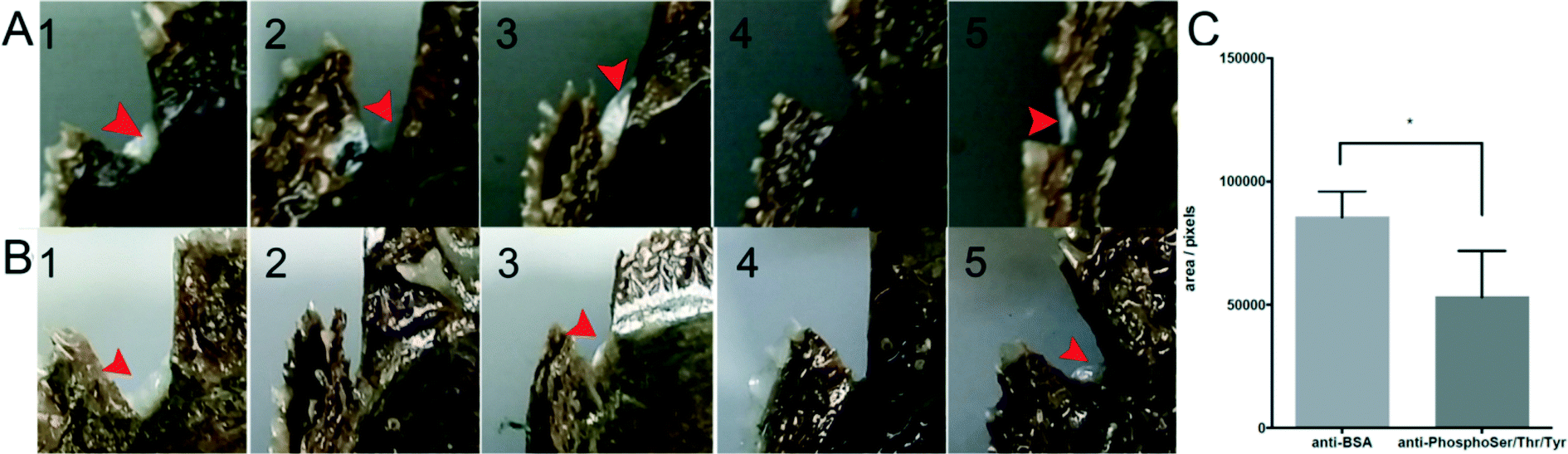 | ||
| Fig. 4 Regenerated shells after shell notching. The regenerated shells of 5 anti-BSA injected oysters (A) and 5 anti-phosphoserine/threonine/tyrosine injected oysters (B) were photographed. The red arrow indicated a nascent shell. And the pixel area of regenerated shells was measured using Photoshop (C). The star represented that there was a significant difference between the two groups (*p < 0.05, analyzed by multiple t-tests). | ||
Phosphorylation of shell matrix proteins is critical for shell formation: in vitro experiment
Next, to examine the role of phosphorylation of SMPs in shell formation in vitro, lambda protein phosphatase was used to remove phosphate groups of SMPs extracted from the shells of P.fucata, which are composed of calcite as the outer prismatic layer and aragonite as the inner nacreous layer. Finally, four groups of proteins were obtained, i.e., an EDTA soluble matrix of prism/nacre (ESM-P/N) and an EDTA insoluble matrix of prism/nacre (EISM-P/N). Then, an enzyme linked immunosorbent assay (ELISA) experiment was conducted to examine the extent of removed phosphate groups in SMPs. It is found that all four matrix proteins groups were dephosphorylated to some extent (Fig. S6†). The A450 absorbance values represented the phosphorylation extent. It is noted that after treatment, the A450 values of the treated groups did not decline to zero, which could be incomplete dephosphorylation. Based on one-way ANOVA analysis, the phosphorylation extent of ESM-P, EISM-N and EISM-P after protein phosphatase treatment was significantly lower compared to that of the untreated SMPs. The phosphorylation extent of ESM-N after protein phosphatase treatment had no significant difference compared to that of the untreated SMPs, which may be due to its low phosphorylation level. Because the A450 of untreated ESM-N (∼0.03) was higher than that of treated ESM-N (∼0.01), it is considered that ESM-N after treatment was also dephosphorylated. From the results, it can be seen that the phosphorylation of SMPs in the EISM was higher than that in the ESM and the phosphorylation of SMPs in the prism was higher than that in the nacre. Since the prism is composed of calcite, the effect of phosphorylation on calcite crystallization needs to be studied.Therefore, to investigate the function of phosphorylation of SMPs, a calcium carbonate crystallization experiment in vitro (calcite reaction systems) was conducted to compare the effect of untreated SMPs and dephosphorylated SMPs. For the control, in the presence of 20 μg mL−1 BSA or H2O, typical rhombohedral calcium carbonate crystals with diameters of 2.5–4.5 μm were produced (Fig. S7†). By contrast, in the presence of 10 μg mL−1 ESM-N (Fig. 5A1 and A2) or ESM-P (Fig. 5C1 and C2), calcium carbonate with shape modification was formed although the sizes of crystals were not significantly changed. It is assumed that the inhibition of calcite steps is due to step-specific binding affinity that results in shape modification, similar to the effect of otoconial proteins on the calcite crystallization.39 Strikingly, in the presence of 10 μg mL−1 dephosphorylated proteins, the shape modification effects on crystals were largely neutralized (Fig. 5B1, B2, D1 and D2). The formed crystals resembled rhombohedral calcium carbonate with slightly modified surfaces (Fig. 5B2) or rounded edges (Fig 5D2). In the presence of 10 μg mL−1 EISM-N (Fig. 6A1 and A2) or EISM-P (Fig. 5C1 and C2), the shape modification of crystals was clear but was different with that in the ESM groups. Step patterns on the faces of the crystals indicated rounding of one certain calcite step and hindering the calcium or carbonate ions from attaching to the kink sites, which is usually associated with preferential binding of proteins to those steps.39 Then, after dephosphorylation, the shape modification ability of the SMPs was compromised, as is evident from the rhombohedral calcium carbonate formed in the presence of dephosphorylated EISMs (Fig. 6B1, B2, D1 and D2). This finding is quite interesting and shows that phosphorylation of SMPs is essential for regulating the morphology of calcium carbonate in vitro, which could be related to the early crystallization process.
 | ||
| Fig. 5 The effect of phosphorylation of the EDTA soluble matrix (ESM) on the morphology of calcium carbonate. SEM images of calcium carbonate crystals in the presence of 10 μg mL−1 untreated ESM (A1, A2, C1, C2) and 10 μg mL−1 dephosphorylated ESM (B1, B2, D1, D2), respectively. ESM-N-DP and ESM-P-DP represented dephosphorylated ESM-N and ESM-P, respectively. | ||
 | ||
| Fig. 6 The effect of phosphorylation of the EDTA insoluble matrix (EISM) on the morphology of calcium carbonate. SEM images of calcium carbonate crystals in the presence of 10 μg mL−1 untreated EISM (A1, A2, C1 and C2) and 10 μg mL−1 dephosphorylated EISM (B1, B2, D1 and D2), respectively. EISM-N-DP and EISM-P-DP represented dephosphorylated EISM-N and EISM-P, respectively. | ||
Next, a calcium carbonate precipitation assay was conducted to investigate the effect of phosphorylation on the rate of calcium carbonate precipitation. The absorbance values at 570 nm (A570) are positively correlated with the amounts of calcium carbonate formed. During five minutes of the reaction, compared with the control group (Fig. 7), all the SMPs had an inhibiting effect on calcium carbonate precipitation. Generally, ESMs (Fig. 7A) inhibited calcium carbonate precipitation more seriously than EISMs (Fig. 7B). At the same time, dephosphorylation alleviated the inhibiting effect of both ESM-N and ESM-P groups (Fig. 7A). Dephosphorylation of EISMs had a less obvious inhibiting effect on crystallization compared to that of ESMs. Nevertheless, it is concluded that phosphorylation has an inhibiting effect on calcium carbonate precipitation. This finding is in agreement with previous studies that the phosphorylated shell matrix from the bivalve shells was more effective in inhibiting crystal growth than the dephosphorylated matrix,23 showing that phosphorylation of SMPs delayed the crystal nucleation process. In addition, the results indicate that the extent of phosphorylation of SMPs is not proportional to their inhibition ability; although the phosphorylation extent of EISMs is generally higher than that of ESMs (Fig. S6†), the latter has more inhibiting effect on calcium carbonate crystallization (Fig. 7).
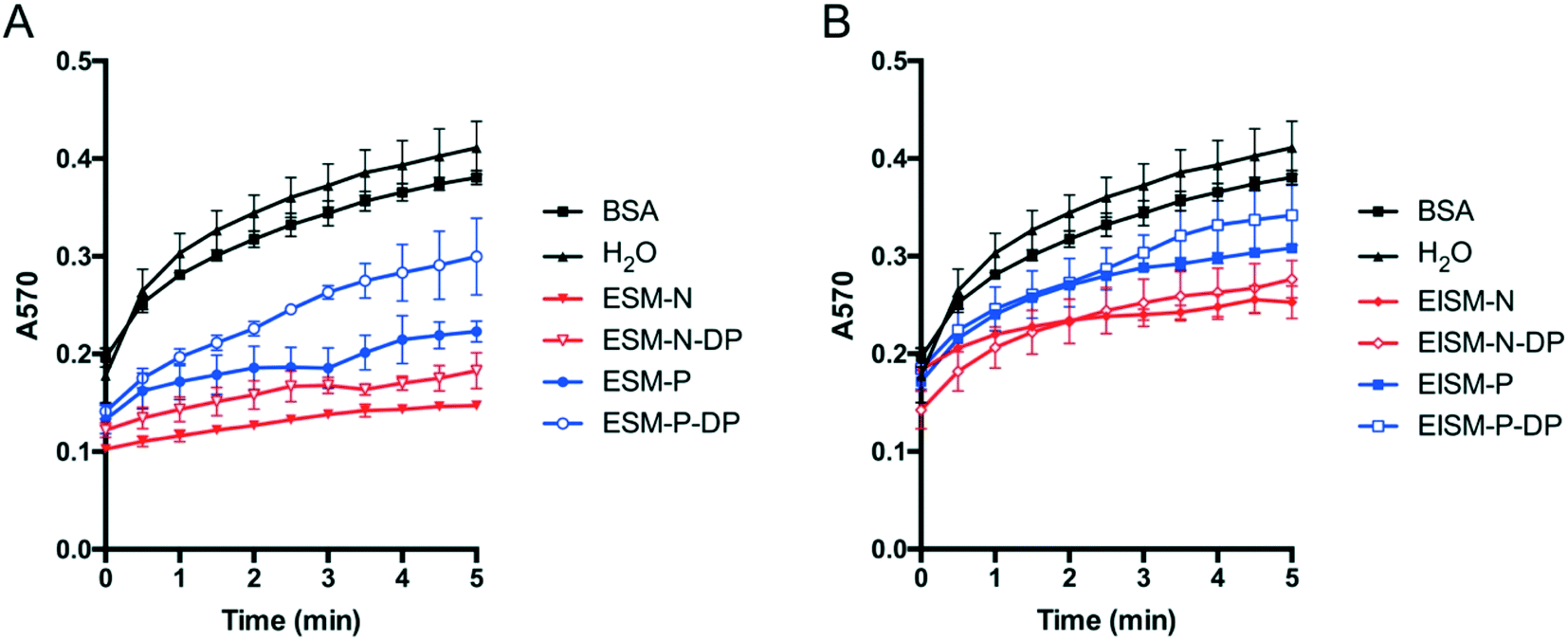 | ||
| Fig. 7 The effect of phosphorylation of SMPs on the calcium carbonate precipitation rate. The A570 absorbance value represented the calcium carbonate precipitation extent. 20 μg mL−1 BSA in water were used as a negative control. The concentration of all untreated SMPs and dephosphorylated SMPs was 10 μg mL−1. ESM (A) and EISM (B) could inhibit calcium carbonate precipitation compared with the negative control. Dephosphorylation of ESM (A) and EISM (B) alleviated the inhibition of precipitation. Error bars, standard deviation of three independent experiments. | ||
Based on the experiments above, it is clear that phosphorylation influences the morphology and the precipitation rate of calcium carbonate. However, the mechanism of the phosphorylation effect remains unclear. A probable explanation is that phosphorylation affects the occlusion of SMPs in crystals, thus affecting calcium carbonate crystal formation. To verify this hypothesis, we labeled ESM proteins with Cy5 and observed the fluorescence distribution in crystals using confocal microscopy. The fluorescence distribution reflected the distribution and occlusion of proteins inside the crystals. The fluorescence intensity was measured to quantify the amounts of protein occlusion. By comparing ESM-N (Fig. 8A) and dephosphorylated ESM-N (Fig. 8B), it was found that the proteins were distributed most in the outer area of crystals when dephosphorylated, and the gray value distributed in the outer area was three times higher than that at the center (Fig. 8B4). By contrast, untreated proteins were distributed both in the outer area and the inner area and the gray value in outer area was only 1.5 times higher than that in the inner area (Fig 8A4). Similar results were observed in the ESM-P groups (Fig. 8C and D). The gray value distributed in the outer area was ten times higher than that at the center in the dephosphorylated ESM-P group (Fig. 8D4) while the gray value in the outer area was only 1.5 times higher than that in the inner area in the untreated group (Fig. 8C4). Moreover, the total amounts of protein occlusions inside the crystals were significantly decreased in the dephosphorylation groups (Fig. 9). It is noted that dephosphorylated ESM-N reduced protein occlusions much more compared to the dephosphorylated ESM-P group. To quantify the amount of occluded proteins, the crystals were washed to remove adsorbed proteins, dissolved in EDTA and then the amount of proteins left after crystal dissolution was measured (Fig. S8†). It can be seen that after dephosphorylation, ESM-N and ESM-P were less occluded in the crystals. The amount of phosphorylated proteins occluded was 2–2.5 times compared to the dephosphorylated proteins. This result supported the fluorescence data in Fig. 8 and 9.
 | ||
| Fig. 8 The occlusion of dephosphorylated SMPs in crystals observed by confocal microscopy. Confocal imaging of calcium carbonate crystals in the presence of 10 μg mL−1 Cy5-labeled untreated ESM-N (A1–A3), dephosphorylated ESM-N (B1–B3), untreated ESM-P (C1–C3) and dephosphorylated ESM-P (D1-D3). Images were taken at 647 nm laser excitation (A1, B1, C1 and D1) and at bright field (A2, B2, C2 and D2). Merged images are A3, B3, C3 and D3. The fluorescence of labeled proteins was measured along the yellow lines marked in A1, B1, C1 and D1. The gray values of fluorescence are shown in A4, B4, C4 and D4. Dephosphorylation reduced the protein occlusion inside the calcium carbonate crystals. | ||
 | ||
| Fig. 9 The fluorescence intensity of SMPs distributed in crystals. The integral intensity represented the fluorescence intensity. The fluorescence intensity of two dephosphorylated groups were significantly lower (** p < 0.01, respectively, p < 0.001 and p = 0.002, analyzed by multiple t-tests) than untreated groups. Error bars, standard deviation of 8–10 measured crystals. | ||
Discussion
The injection of antibodies against the anti-phosphoserine/threonine/tyrosine sites of matrix proteins greatly declined the phosphorylation and caused abnormal growth both in the prismatic and nacreous layers. It seems that decreased phosphorylation causes more abnormal growth of nacre tablets. Interestingly, this overgrowth of the nacre pattern is similar to the effect of Fam20C knockdown;25 again supporting that Fam20C participated in the shell formation through phosphorylating matrix proteins. The holes in the prismatic layers resemble the effect of KRMP8 and Alv-like knockdown (unpublished data), suggesting that the phosphorylation of the two matrix proteins contributed more to the formation of the prismatic layer. However, the mechanisms of crystal growth distortion caused by antibody injection or RNA interference are still unknown. The Raman spectra especially on the nacre showed the reduced crystallinity and crystal orientation change after antibody injection. The regeneration of shells after shell notching is a unique model for studying shell formation. During shell regeneration, many matrix proteins were upregulated.8 It is found that the regeneration of shells was slowed down, indicating that the phosphorylation of matrix proteins is critical for shell regeneration. Previously, it is shown that the SM fraction from foliated calcitic shells is 12.9–15.7% phosphate by weight while SM fractions isolated from other calcitic prismatic or various aragonitic microstructures contain 0–3.3% phosphate by weight, suggesting that the phosphorylation level is related to the structural difference of shells.23 Together, these in vivo studies show direct evidence that the formation of both the prismatic layer and nacre layer relies on the phosphorylation of matrix proteins in the extrapallial fluid.Besides phosphorylation, glycosylation is another form of protein modification. However, glycosylation of EISM and ESMs of P.fucata shells and their effects on mineralization are currently unknown. Previously, glycosylation of shell matrix proteins have been largely neglected and most of the studies simply predicted the glycosylation status in SMPs such as Asprich40 and Lustrin A.41 Recently, Eric P. Chang et al. have investigated the effect of glycosylation of AP24 protein on CaCO3 mineralization and found that glycosylation of AP24 has more inhibition on nucleation and crystal modification compared to unglycosylated AP24.42
Through the in vitro experiment, it is found that the calcium carbonate morphology modulating ability was largely compromised for dephosphorylated SMPs in all the four fractions of proteins extracted from the shells. The results were quite surprising because the phosphorylation extent of proteins is still half of the control. Phosphorylation of OPN was required for the inhibition of vascular smooth muscle cell calcification and inhibition of calcium phosphate20 and calcium oxalate crystallization.21 Besides inhibition of mineral crystallization, phosphorylation of proteins affected the collagen assembly, an important player in vertebrate biomineralization. Phosphorylated dentin matrix protein 1 and dentin phosphophoryn (DPP) facilitated organized mineralization of collagen fibrils.43 DPP could nucleate plate-like apatite crystals whereas rDMP2 failed to mediate the transformation of amorphous calcium phosphate to apatite crystals.44 In shell formation, a “chitin-silk fibroin gel protein-acidic macromolecules” mode was proposed before,45 thus, the chitin and silk fibroin may also interact with phosphorylated SMPs, which need further studies. From the perspective of calcium binding, it was found that phosphorylation of orchestin, a major matrix protein from crustacean, was necessary for calcium binding of the protein and calcium binding only occurs via the phosphoserine residues.46 Similarly, the affinity of the starmaker protein, a protein controlling otolith morphogenesis in otolith morphogenesis,2 for calcium binding was dependent on its phosphorylation state.47 From the perspective of crystal growth, phosphorylated OPN preferentially bound to the [![[1 with combining overline]](https://www.rsc.org/images/entities/char_0031_0305.gif) 00]Cc steps, which may alter mineral interfacial energies, thus delaying the formation of active steps during brushite growth. Furthermore, the phosphorylation-deficient form of OPN failed to inhibit brushite crystallization.20 Piotr Dobryszycki et al. found that the dimensions of the calcite crystals growing in the presence of the phosphorylated starmaker protein were smaller in comparison with the control.48 These studies together with our results show that phosphorylation of proteins affects the crystal nucleation, size, morphology but not the crystal polymorph, which is in agreement with our in vivo studies. Generally, inhibition of calcium carbonate formation is required for the control of shell formation. It may allow shells to grow normally in the presence of other shell matrix proteins. Loss of inhibition of calcium carbonate results in overgrowth and disordered growth of shells, partially shown in the experiment in vivo (Fig. 1 and 2).
00]Cc steps, which may alter mineral interfacial energies, thus delaying the formation of active steps during brushite growth. Furthermore, the phosphorylation-deficient form of OPN failed to inhibit brushite crystallization.20 Piotr Dobryszycki et al. found that the dimensions of the calcite crystals growing in the presence of the phosphorylated starmaker protein were smaller in comparison with the control.48 These studies together with our results show that phosphorylation of proteins affects the crystal nucleation, size, morphology but not the crystal polymorph, which is in agreement with our in vivo studies. Generally, inhibition of calcium carbonate formation is required for the control of shell formation. It may allow shells to grow normally in the presence of other shell matrix proteins. Loss of inhibition of calcium carbonate results in overgrowth and disordered growth of shells, partially shown in the experiment in vivo (Fig. 1 and 2).
It is common to find biomacromolecules occluded in biogenic crystals, such as those found in mollusk shells.49,50 For example, prisms from the calcitic layer of mollusk shells are single crystals of calcite with an incorporated organic matrix composed of proteins and other biomacromolecules.27 The occlusion of biomacromolecules within individual crystals is highly relevant to their superior mechanical properties.51 Organic occlusions could add additional energy dissipation mechanisms at the nanoscale and microscale.52 Interestingly, in dental mineralization, amelogenin processing by matrix metalloproteinase 20 prevents protein occlusion inside calcite crystals53 and apatite crystals,54 in which full-length amelogenin may bind most strongly, truncated amelogenin binds weakly, and the C-terminus alone has the weakest interaction to hydroxyapatite and calcite. The occlusion of organics in enamel undermine the mechanical properties of biominerals while in shells, organic occlusions enhance the hardness and stiffness of the materials.55,56 In this study, we found that protein occlusion inside calcium carbonate was greatly reduced in dephosphorylated ESM groups compared to the phosphorylated groups, indicating that phosphorylation of SMPs was required for protein occlusion inside the crystals. This could be due to the phosphorylated groups with negative charges that can attract Ca2+ at the early stages of crystallization and facilitate the interaction between proteins and crystals. It is expected that phosphorylated proteins stick to growing calcite crystals and consequently modify their morphology, because the phosphate groups bind with the calcium ions on the surface of the growing crystals, and thereby inhibit crystal growth where they bind. It is also expected, therefore, that when the phosphate groups are removed the protein molecules will bind less to calcium, and consequently will interfere less with crystal growth. Therefore, the phosphorylated proteins are found in greater amounts inside the crystals, and are found also in the core of the crystals, whereas the less phosphorylated molecules crowd at the crystal surface. The binding of phosphorylated proteins onto the surfaces of the crystal is also possible. But after PBS and DI water washing, we think that this portion is limited. Last but not the least, the phosphorylated SMPs could also be related to amorphous calcium carbonate (ACC) stabilization57 or transformation,58 and need further investigation. Interestingly, occlusion of more proteins should decrease the crystallinity rather than increase it. Currently, the explanation for this phenomenon is unknown. We speculate that it may be related to the status of proteins occluded in the unit cell of crystals, which need further studies. In addition, knowledge of the forms of phosphorylated proteins occluded in vivo is required to address the questions.
Conclusions
Through both in vivo and in vitro experiments, we demonstrated that phosphorylation of matrix proteins is critical for shell formation. Reduction of phosphorylation resulted in abnormal shell formation but without affecting the crystal polymorph and inhibited shell regeneration in vivo. In vitro reduction of phosphorylation caused a large loss of function of matrix proteins on modulation of the crystal morphology and reduced protein occlusion inside the crystals.Author contributions
R. Z., J. Z. D. and C. L. conceived and designed the project. J. Z. D., C. L. and G. X. carried out the experiments. The manuscript was written by C. L. and J. Z. D.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China, Grants 31372502 and 31572594. Open Access funding provided by the Max Planck Society.References
- H. A. Lowenstam, S. Weiner and S. Weiner, On biomineralization, Oxford University PressNew York, 1989 Search PubMed.
- C. Sollner, M. Burghammer, E. Busch-Nentwich, J. Berger, H. Schwarz, C. Riekel and T. Nicolson, Science, 2003, 302, 282–286 CrossRef PubMed.
- B. Marie, C. Joubert, A. Tayalé, I. Zanella-Cléon, C. Belliard, D. Piquemal, N. Cochennec-Laureau, F. Marin, Y. Gueguen and C. Montagnani, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 20986–20991 CrossRef PubMed.
- M. A. Hood, H. Leemreize, A. Scheffel and D. Faivre, J. Struct. Biol., 2016, 196, 147–154 CrossRef PubMed.
- C. Liu, S. Li, J. Kong, Y. Liu, T. Wang, L. Xie and R. Zhang, Sci. Rep., 2015, 5, 17269 CrossRef PubMed.
- P. Fratzl and R. Weinkamer, Prog. Mater. Sci., 2007, 52, 1263–1334 CrossRef.
- A. M. Belcher, X. Wu, R. Christensen, P. Hansma, G. Stucky and D. Morse, Nature, 1996, 381, 56–58 CrossRef.
- D. Fang, G. Xu, Y. Hu, C. Pan, L. Xie and R. Zhang, PLoS One, 2011, 6, e21860 CrossRef PubMed.
- D. Fang, C. Pan, H. Lin, Y. Lin, G. Zhang, H. Wang, M. He, L. Xie and R. Zhang, J. Biol. Chem., 2012, 287, 15776–15785 CrossRef PubMed.
- C. Pan, D. Fang, G. Xu, J. Liang, G. Zhang, H. Wang, L. Xie and R. Zhang, J. Biol. Chem., 2014, 289, 2687–2776 CrossRef PubMed.
- F. F. Amos, E. Destine, C. B. Ponce and J. S. Evans, Cryst. Growth Des., 2010, 10, 4211–4216 CrossRef.
- C. B. Ponce and J. S. Evans, Cryst. Growth Des., 2011, 11, 4690–4696 CrossRef.
- E. C. Keene, J. S. Evans and L. A. Estroff, Cryst. Growth Des., 2010, 10, 1383–1389 CrossRef.
- S. Y. Bahn, B. H. Jo, Y. S. Choi and H. J. Cha, Sci. Adv., 2017, 3, e1700765 CrossRef PubMed.
- F. Nudelman, B. A. Gotliv, L. Addadi and S. Weiner, J. Struct. Biol., 2006, 153, 176–187 CrossRef PubMed.
- A. Veis, C. Sfeir and C. B. Wu, Crit. Rev. Oral Biol. Med., 1997, 8, 360–379 CrossRef.
- A. George and A. Veis, Chem. Rev., 2008, 108, 4670–4693 CrossRef PubMed.
- V. S. Tagliabracci, J. L. Engel, J. Wen, S. E. Wiley, C. A. Worby, L. N. Kinch, J. Xiao, N. V. Grishin and J. E. Dixon, Science, 2012, 336, 1150–1153 CrossRef PubMed.
- S. Jono, C. Peinado and C. M. Giachelli, J. Biol. Chem., 2000, 275, 20197–20203 CrossRef PubMed.
- S. Li, S. Wu, D. Nan, W. Zhang and L. Wang, Chem. Mater., 2014, 26, 5605–5612 CrossRef.
- L. Wang, X. Guan, R. Tang, J. R. Hoyer, A. Wierzbicki, J. J. De Yoreo and G. H. Nancollas, J. Phys. Chem. B, 2008, 112, 9151–9157 CrossRef PubMed.
- Y. C. Chien, M. T. Hincke, H. Vali and M. D. McKee, J. Struct. Biol., 2008, 163, 84–99 CrossRef PubMed.
- J. E. Borbas, A. P. Wheeler and C. S. Sikes, J. Exp. Zool., 1991, 258, 1–13 CrossRef.
- K. Mann and E. Edsinger, Proteome Sci., 2014, 12, 28 CrossRef PubMed.
- J. Du, C. Liu, G. Xu, J. Xie, L. Xie and R. Zhang, Sci. Rep., 2018, 8, 3563 CrossRef PubMed.
- K. Gries, R. Kröger, C. Kübel, M. Fritz and A. Rosenauer, Acta Biomater., 2009, 5, 3038–3044 CrossRef PubMed.
- H. Li, H. L. Xin, M. E. Kunitake, E. C. Keene, D. A. Muller and L. A. Estroff, Adv. Funct. Mater., 2011, 21, 2028–2034 CrossRef.
- S. Younis, Y. Kauffmann, L. Bloch and E. Zolotoyabko, Cryst. Growth Des., 2012, 12, 4574–4579 CrossRef.
- E. Weber and B. Pokroy, CrystEngComm, 2015, 17, 5873–5883 RSC.
- Y.-Y. Kim, J. D. Carloni, B. Demarchi, D. Sparks, D. G. Reid, M. E. Kunitake, C. C. Tang, M. J. Duer, C. L. Freeman, B. Pokroy, K. Penkman, J. H. Harding, L. A. Estroff, S. P. Baker and F. C. Meldrum, Nat. Mater., 2016, 15, 903–910 CrossRef PubMed.
- A. S. Mount, A. P. Wheeler, R. P. Paradkar and D. Snider, Science, 2004, 304, 297–300 CrossRef PubMed.
- C. Liu, L. Xie and R. Zhang, Sci. Rep., 2015, 5, 18338 CrossRef PubMed.
- M. Suzuki, E. Murayama, H. Inoue, N. Ozaki, H. Tohse, T. Kogure and H. Nagasawa, Biochem. J., 2004, 382, 205–213 CrossRef PubMed.
- N. Blom, S. Gammeltoft and S. Brunak, J. Mol. Biol., 1999, 294, 1351–1362 CrossRef PubMed.
- M. Suzuki, K. Saruwatari, T. Kogure, Y. Yamamoto, T. Nishimura, T. Kato and H. Nagasawa, Science, 2009, 325, 1388–1390 CrossRef PubMed.
- J. Xie, J. Liang, J. Sun, J. Gao, S. Zhang, Y. Liu, L. Xie and R. Zhang, Cryst. Growth Des., 2016, 16, 672–680 CrossRef.
- C. Liu, G. Xu, J. Du, J. Sun, X. Wan, X. Liu, J. Su, J. Liang, G. Zheng, L. Xie and R. Zhang, Cryst. Growth Des., 2018, 18, 32–36 CrossRef.
- K. Ramesh, F. Melzner, A. W. Griffith, C. J. Gobler, C. Rouger, D. Tasdemir and G. Nehrke, J. R. Soc., Interface, 2018, 15, 20170723 CrossRef PubMed.
- M. Hong, T. K. Moreland, J. Chen, H. H. Teng, R. Thalmann and J. J. De Yoreo, Cryst. Growth Des., 2015, 15(1), 129–136 CrossRef PubMed.
- B. A. Gotliv, N. Kessler, J. L. Sumerel, D. E. Morse, N. Tuross, L. Addadi and S. Weiner, ChemBioChem, 2005, 6, 304–314 CrossRef PubMed.
- X. Shen, A. M. Belcher, P. K. Hansma, G. D. Stucky and D. E. Morse, J. Biol. Chem., 1997, 272, 32472–32481 CrossRef PubMed.
- E. P. Chang, I. Perovic, A. Rao, H. Cölfen and J. S. Evans, Biochemistry, 2016, 55, 1024–1035 CrossRef PubMed.
- A. S. Deshpande, P.-A. Fang, X. Zhang, T. Jayaraman, C. Sfeir and E. Beniash, Biomacromolecules, 2011, 12, 2933–2945 CrossRef PubMed.
- G. He, A. Ramachandran, T. Dahl, S. George, D. Schultz, D. Cookson, A. Veis and A. George, J. Biol. Chem., 2005, 280, 33109–33114 CrossRef PubMed.
- L. Addadi, D. Joester, F. Nudelman and S. Weiner, Chem. – Eur. J., 2006, 12, 980–987 CrossRef PubMed.
- A. Hecker, O. Testenière, F. Marin and G. Luquet, FEBS Lett., 2003, 535, 49–54 CrossRef PubMed.
- M. Wojtas, R. Hołubowicz, M. Poznar, M. Maciejewska, A. Ożyhar and P. Dobryszycki, Biochemistry, 2015, 54, 6525–6534 CrossRef PubMed.
- M. Wojtas, M. Wołcyrz, A. Ożyhar and P. Dobryszycki, Cryst. Growth Des., 2011, 12, 158–168 CrossRef.
- B. Pokroy, J. P. Quintana, E. N. Caspi, A. Berner and E. Zolotoyabko, Nat. Mater., 2004, 3, 900–902 CrossRef PubMed.
- B. Pokroy, A. N. Fitch, F. Marin, M. Kapon, N. Adir and E. Zolotoyabko, J. Struct. Biol., 2006, 155, 96–103 CrossRef PubMed.
- K. Rae Cho, Y. Y. Kim, P. Yang, W. Cai, H. Pan, A. N. Kulak, J. L. Lau, P. Kulshreshtha, S. P. Armes, F. C. Meldrum and J. J. De Yoreo, Nat. Commun., 2016, 7, 10187 CrossRef PubMed.
- C. Liu, J. Du, L. Xie and R. Zhang, Cryst. Growth Des., 2017, 17, 1966–1976 CrossRef.
- K. M. Bromley, R. Lakshminarayanan, M. Thompson, S. B. Lokappa, V. A. Gallon, K. R. Cho, S. R. Qiu and J. Moradian-Oldak, Cryst. Growth Des., 2012, 12, 4897–4905 CrossRef PubMed.
- S. Prajapati, J. Tao, Q. Ruan, J. J. De Yoreo and J. Moradian-Oldak, Biomaterials, 2016, 75, 260–270 CrossRef PubMed.
- S. Weiner and L. Addadi, J. Mater. Chem., 1997, 7, 689–702 RSC.
- L. Li, J. C. Weaver and C. Ortiz, Nat. Commun., 2015, 6, 6216 CrossRef PubMed.
- S. Bentov, S. Weil, L. Glazer, A. Sagi and A. Berman, J. Struct. Biol., 2010, 171, 207–215 CrossRef PubMed.
- S. Kababya, A. Gal, K. Kahil, S. Weiner, L. Addadi and A. Schmidt, J. Am. Chem. Soc., 2015, 137, 990–998 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ce00755a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |