
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntensified deracemization via rapid microwave-assisted temperature cycling†
Fabio
Cameli‡
,
Christos
Xiouras‡
 and
Georgios D.
Stefanidis
*
and
Georgios D.
Stefanidis
*
Process Engineering for Sustainable Systems (ProcESS), Department of Chemical Engineering KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium. E-mail: Georgios.Stefanidis@kuleuven.be
First published on 16th May 2018
Abstract
Complete solid-state deracemization can be achieved via microwave-assisted temperature cycling. The combined application of rapid cooling and microwave heating could drastically reduce both process time and the impact of side reactions that lower productivity. While operating at this novel process window induces secondary nucleation, deracemization per cycle is not hindered.
Single enantiomers are ubiquitous in bio-systems, yet their chemical synthesis from achiral or racemic precursors presents a major challenge.1 Current approaches in asymmetric or enantioselective synthesis starting from achiral compounds heavily rely on the use of chiral auxiliaries or asymmetric (bio)-catalysts.2 However, “true” chiral symmetry breaking processes without external agents, such as the Soai reaction,2b have also been demonstrated. In these cases, each enantiomer acts as a catalyst for its own formation (asymmetric autocatalysis) leading to amplification of a very small initial enantiomeric excess that is stochastically formed. Such examples of absolute asymmetric synthesis are, however, rare in solution phase chemistry.2c
Chiral symmetry breaking phenomena are observed more frequently involving the solid state, which may exhibit much stronger chiral discrimination, i.e. crystallization as a racemic mixture of enantiopure crystals (racemic conglomerate).3 An example of such phenomenon is Viedma ripening,4 in which total deracemization is obtained by grinding a slurry of chiral crystals, which undergo size-dependent dissolution and racemize in the liquid phase. Viedma ripening was initially demonstrated for the achiral NaClO3 that forms chiral conglomerate crystals,4a but the method was later applied to several chiral compounds, such as amino acids4e–f and their derivatives,4g–i pharmaceuticals4j and organometallic complexes.4k A recent study also revealed that Viedma ripening can be combined with a reversible reaction between achiral substrates to deliver enantiopure solids, in an elegant example of an absolute asymmetric synthesis.4l However, while Viedma ripening is promising for practical application, scale up of the process is complicated, because the intense grinding required to achieve fast deracemization rate necessitates the use of ball/bead mills or similar energy intensive equipment. In addition, further processing steps may be required to separate the grinding media from the solid product. The application of alternative energy transfer mechanisms in deracemization has been discussed as a potential means to enhance the process and address some of the aforementioned limitations, with the recent application of ultrasound grinding as a prominent example.5
Besides grinding, the application of temperature gradients6 or cycles7 can also lead to complete deracemization. In the latter approach, programmed heating and cooling cycles are repeatedly applied in the suspension. During the heating step, the enantiomeric crystals in the solid phase partially dissolve, while in the low temperature period, crystallization takes place in favour of the excess enantiomer. The solution racemization reaction guarantees a racemic liquid composition that limits the nucleation of the minor species, thus maintaining the enrichment towards the major enantiomer.7a
The impact of several process parameters, such as initial enantiomeric excess, amount of solid and catalyst, operating volume, temperature swings amplitude and cooling rate have been studied experimentally7a–e and numerically.7f–h Regarding the effect of the cooling rate, Suwannasang et al.7a reported that the deracemization efficiency per cycle decreased upon increase in the cooling rate, presumably due to secondary nucleation of the counter enantiomer, but the process was equally fast in time due to the reduction in cycle duration. Similarly, Breveglieri et al.7e showed that while faster cooling decreased the deracemization efficiency per cycle, it allowed for faster process due to the shorter cycles. Conversely, Li et al.7d observed similar deracemization per cycle irrespective of the cooling rate. While these studies all conclude that fast cooling may shorten the overall process time, the cooling rates applied so far are still rather low (0.15–1.3 °C min−1). In addition, although the effect of the heating rate has not been studied yet, a similar effect in reducing the total process time is expected for fast heating rates as well. Therefore, it is reasonable to assume that the extreme operating window for fastest deracemization may be that of instantaneous cooling and heating steps followed by (optimized) isothermal holding steps at the desired temperatures.
Herein, we perform deracemization of the model compound glutamic acid by investigating novel operating windows: the ramps between the two temperatures are eliminated by carrying out the process in a microwave reactor, which enables instantaneous heating (20 °C min−1) and very fast cooling (16.7 °C min−1). The elimination of the heating and cooling steps reduces the cycle duration (Fig. 1) and effectively allows the dissolution and crystallization steps to proceed isothermally for the first time.
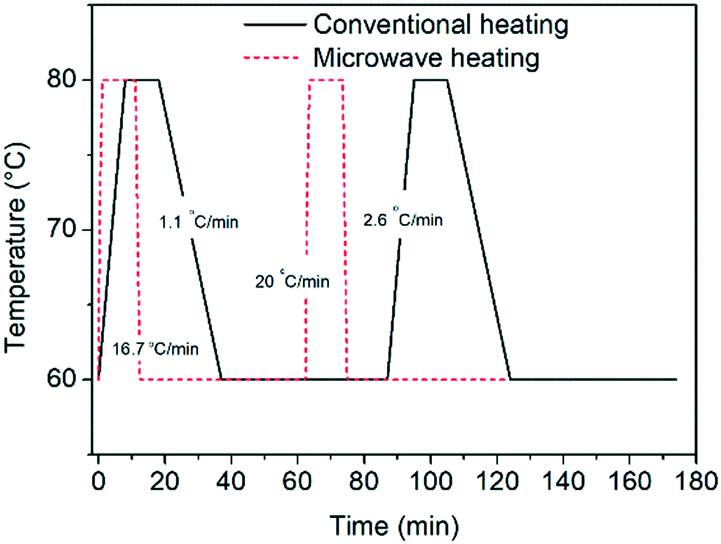 | ||
| Fig. 1 Temperature profiles attained in a microwave reactor equipped with forced air cooling and a conventionally heated jacketed reactor. | ||
Glutamic acid belongs to the family of the so-called “kinetic conglomerates”, since this crystal form is metastable with respect to the anhydrous racemic compound, but the conglomerate form can survive long enough for deracemization to occur.4f,8 Although deracemization of glutamic acid has been observed isothermally starting from a mixture of racemic hydrate crystals that convert to conglomerates,4f,8 to our knowledge, no study has yet reported its deracemization starting from conglomerate crystals of both chiralities. Glutamic acid can be racemized in acidic media at temperatures between 60–100 °C (Fig. 2) using catalytic amounts of aldehydes (e.g. salicylaldehyde).8 However, at such elevated temperatures, parallel to the racemization reaction, decomposition reactions can occur, depleting part of the reagent during the process. In particular, dehydration of glutamic acid into pyroglutamic acid has been observed in acidic media at temperatures >70 °C.4f Based on the solubility data of glutamic acid (ESI†) in acetic acid, temperature cycles between 60–80 °C were applied to allow fast racemization at the lowest temperature and to significantly prevent the decomposition reaction at the upper limit. For such thermolabile compounds, it is desirable to keep the suspension at the two temperatures only for the time necessary for dissolution and crystallization to take place, in order to prevent decomposition.
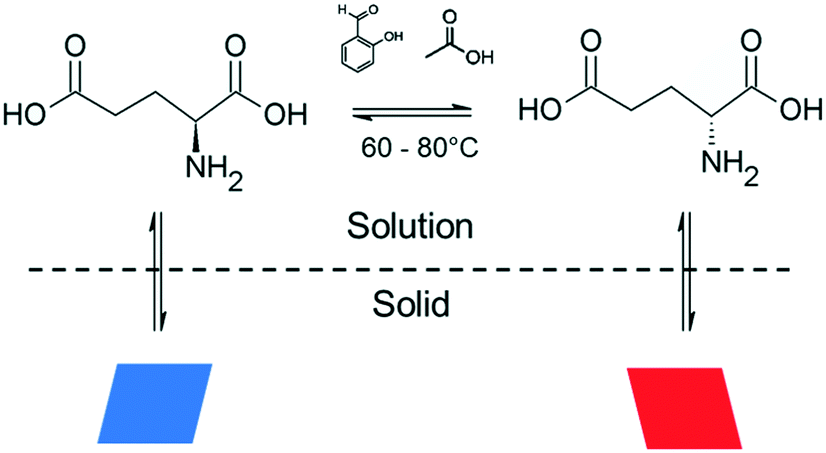 | ||
| Fig. 2 Equilibria involved in glutamic acid deracemization. | ||
In order to test the effect of operating the process at very fast heating and cooling rates, the performance between a microwave reactor equipped with forced air cooling and a jacketed reactor connected to a thermostatic bath which provides hot and cold water was compared. Both configurations work at the same holding times at the low and high temperatures, with the only difference between the profiles being the transient steps of the thermal sweeps in the suspension. The temperature profiles attained are presented in Fig. 1, which reveals that the use of the microwave reactor allows for an order of magnitude faster heating and cooling rates compared to the conventional heating configuration, which in turn leads to ∼30% reduction in the cycle duration. Subsequently, deracemization experiments were conducted using both temperature profiles starting from initial enantiomeric excess values of 22% and 60% in favour of the L enantiomer. Fig. 3 shows that both profiles achieve a similar cycle-based deracemization rate, indicating that fast heating and cooling steps do not substantially influence the deracemization efficiency in a single cycle for the model system studied here. However, Fig. 4 shows that the microwave experiments obtain enantiopurity in a significantly shorter time, mainly due to the reduction in the total cycle duration. Either from Fig. 3 or 4, it is clear that this comparison is unaffected by the initial enantiomeric excess.
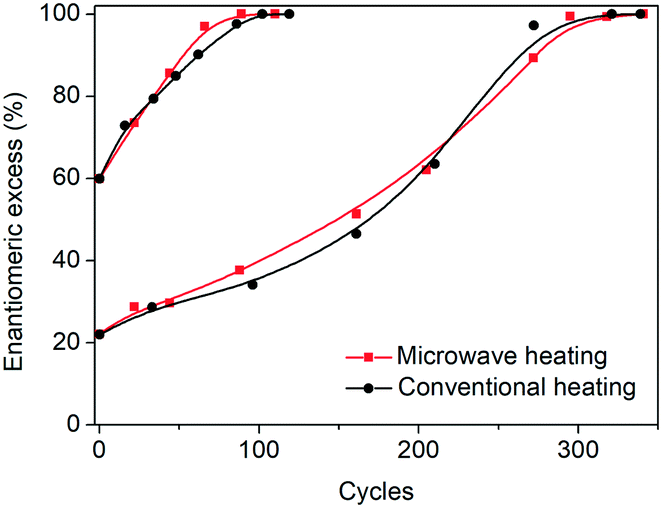 | ||
| Fig. 3 Evolution of e.e. versus number of cycles for microwave and conventional heating configurations. | ||
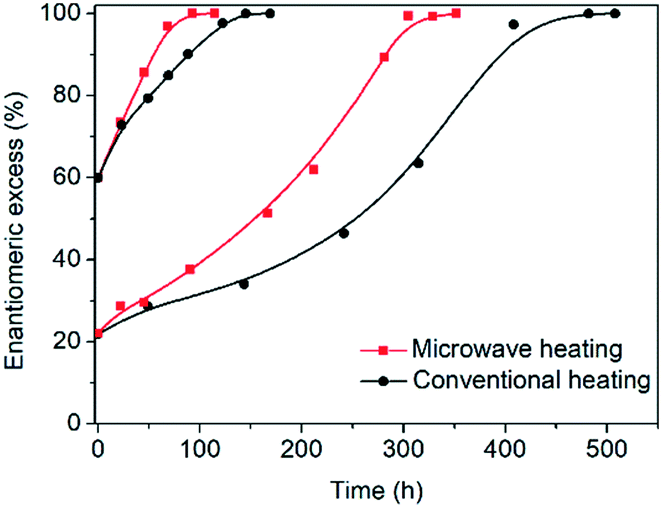 | ||
| Fig. 4 Evolution of e.e. versus time for microwave and conventional heating configurations. | ||
In order to assess the decomposition of glutamic acid in the two experimental configurations, two experiments starting from 60% enantiomeric excess were repeated without intermittent sampling. After 110 cycles, the solid was separated from the liquid and the two phases were analysed. In both cases the solid consisted of enantiopure L-glutamic acid crystals, but in the microwave, the final solid recovered was 87.8% of the initial amount versus 73.9% for the jacketed reactor. The reason for this difference is the slow production of pyroglutamic acid in the liquid phase that induces further dissolution of glutamic acid crystals leading to product loss. Since pyroglutamic acid does not participate in the deracemization process and is more soluble than glutamic acid in this system, it accumulates in the liquid phase. Indeed, in the jacketed reactor, the liquid phase contained 12.5 mg mL−1 of pyroglutamic acid as compared to the microwave reactor in which only 5.5 mg mL−1 was present. Formation of pyroglutamic acid is thus supressed in the microwave reactor, due to the fast heating and cooling steps that allow the mixture to spend less fraction of time at the higher temperatures, where the decomposition reaction progresses faster. This observation is in-line with the selectivity improvements often reported in many reactions, when fast microwave heating replaces slow conventional heating.9 As a result, even though the two experiments achieve complete deracemization at the same number of cycles, in the microwave process 69.6% of the counter enantiomer is converted versus only 35.2% in the conventional heating one. The combination of reduced cycle duration and higher counter enantiomer conversion affords a 3-fold higher productivity of the desired enantiomer crystals in the microwave-assisted process compared to the conventionally heated one (0.064 gL−1 h−1versus 0.023 gL−1 h−1).
In most studies of temperature cycling-enhanced deracemization, the cooling rates applied were deliberately low to allow the relative dominance of crystal growth over secondary nucleation. However, that might not be the case in the operating windows accessible in the microwave reactor. In this configuration, temperature decreases from 80 °C to 60 °C in ∼1 min, creating a supersaturation ratio of ∼4 (ESI†). At such conditions, the possibility of secondary nucleation cannot be excluded. To assess this, we measured the final mean crystal size (D4,3) for the previous experiments. In both cases, smaller particles were obtained in the end of the process: 19.9 μm for the microwave experiment, versus 36 μm for the conventionally heated one. It is noted that the same initial crystals with a mean size of 45.8 μm were used in both experiments. The initial and final mean size, combined with the initial and final total mass allows the approximation of the change in the total number of the particles (Nfin/Nin) during the entire deracemization process (ESI†). For the jacketed reactor, the total number of particles remains roughly unchanged (Nfin/Nin = 1.5), which indicates that growth and dissolution are the dominant phenomena, with the latter being more pronounced, due to the overall smaller particles obtained in the end. Conversely, in the microwave experiments, the number of particles increases substantially (Nfin/Nin = 10.7), which indicates the occurrence of secondary nucleation. However, since similar cycle-based deracemization rates were attained between the two processes, it can be concluded that secondary nucleation does not delay the deracemization process for the system examined here. In fact, secondary nucleation is known to cause enantiomeric enrichment in chiral crystallization,10 due to phenomena of autocatalysis and mutual inhibition.11 Thus, contrary to what is currently believed, for certain compounds, secondary nucleation might also play a substantial role in enabling temperature cycling-enhanced deracemization.
Another key difference in the rapid heating/cooling rates explored here is that contrary to conventional experiments, the dissolution and crystallization steps occur almost exclusively in an isothermal manner at the required temperatures and thus at higher driving forces. As a result, the isothermal periods applied (especially at low temperature) are expected to influence the process. Therefore, we investigated the effect of the holding time at the low temperature on the deracemization capacity per cycle and time, by operating three different temperature profiles using the microwave reactor (Fig. 5). The limiting case is the absence of the isothermal step at 60 °C which, as shown in Fig. 6, does not induce deracemization up to around 400 cycles, when the other two profiles already achieved enantiopurity. Ultimately, using this profile, complete dissolution of the racemic amount in the solid phase was observed due to the production of pyroglutamic acid. However, from the comparison between the profiles with 25 min and 50 min holding times at low temperature (Fig. 6), a slightly higher deracemization capacity per cycle is reached by the longer isothermal stage. On the other hand, by reducing the isothermal step up to 25 min, the process time greatly reduces, thus overcompensating for the lower deracemization capacity per cycle with a higher number of cycles achievable in a shorter time (Fig. 7). Hence, an optimum holding time exists, where maximum deracemization per cycle in the shortest possible time is achieved.
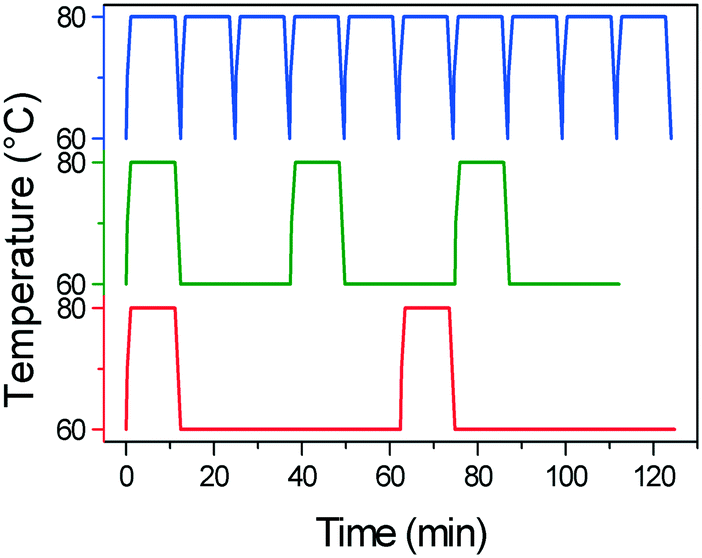 | ||
| Fig. 5 Temperature profiles applied in the microwave experiments with varying holding time at 60 °C: no holding time (blue line), 25 min (green line) and 50 min (red line). | ||
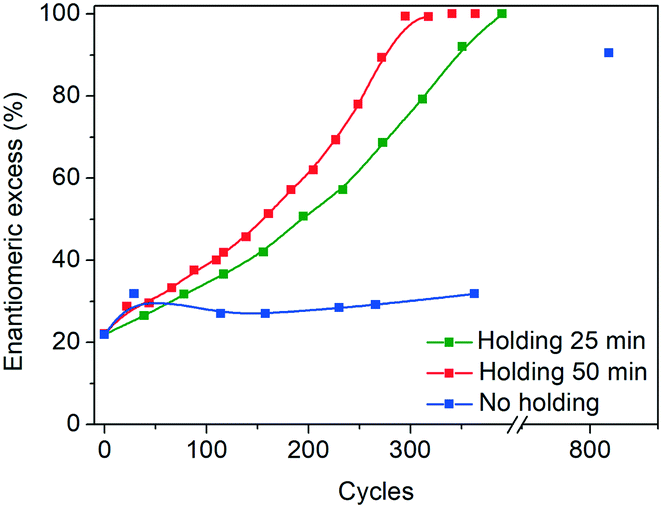 | ||
| Fig. 6 Evolution of e.e. versus number of cycles in the microwave reactor with different holding times at low temperature. | ||
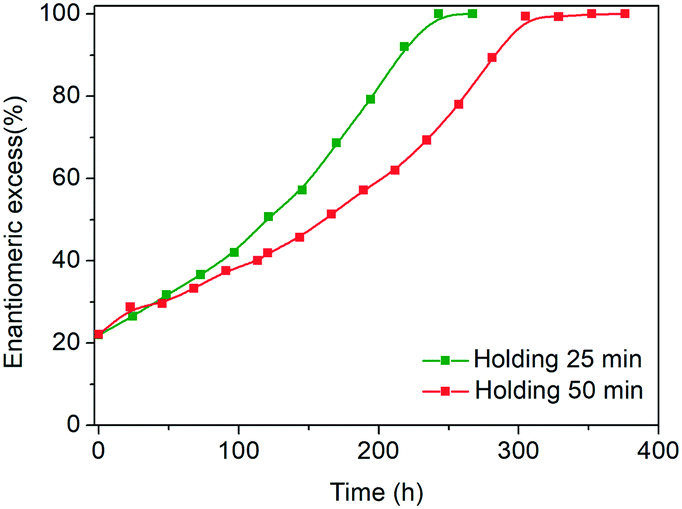 | ||
| Fig. 7 Evolution of e.e. versus time in the microwave reactor with different holding times at low temperature. | ||
In summary, we performed complete temperature cycling-enhanced deracemization of a thermolabile compound using very fast cooling and rapid microwave heating. Operating in these new process windows allowed for much faster deracemization, reduction in side product formation and a 3-fold higher productivity of the preferred enantiomer, as compared to conventional operation. While fast cooling favours secondary nucleation, a similar deracemization per cycle was attained compared to the conventional slow-cooled growth/dissolution-dominated regime, which indicates that secondary nucleation could also enable deracemization. Finally, in the microwave reactor, the isothermal crystallization step (holding time at low temperature) can be optimized to further minimize the process time.
Deracemization experiments: same reactor geometry was used for both MW (Anton Paar Monowave 300) and thermostatic bath (Lauda RE 630) configurations. In all deracemization experiments, 546 mg glutamic acid (e.e. 22% or 60%) were added to 10.5 mL of acetic acid and 120 μL of salicylaldehyde. The suspension was stirred at 600 rpm by an oval PTFE-coated magnetic bar. Suspension samples were taken with a pipette and filtered on a P4 glass filter. The solid phase enantiomeric excess was measured by chiral HPLC (ESI†).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 http://cosmic-etn.eu/. Christos Xiouras acknowledges funding of a Ph.D. fellowship by the Research Foundation-Flanders (FWO).Notes and references
- H. Lorenz and A. Seidel-Morgenstern, Angew. Chem., Int. Ed., 2014, 53, 1218–1250 CrossRef PubMed.
- (a) W. S. Knowles and M. J. Sabacky, Chem. Commun., 1968, 1445 RSC; (b) K. Soai, T. Shibata, H. Morioka and K. Choji, Nature, 1995, 378, 767–768 CrossRef; (c) B. L. Feringa and R. A. Van Delden, Angew. Chem., Int. Ed., 1999, 38, 3418–3438 CrossRef.
- G. Coquerel, in Advances in Organic Crystal Chemistry: Comprehensive Reviews, Springer, 2015, pp. 393–420 Search PubMed.
- (a) C. Viedma, Phys. Rev. Lett., 2005, 94, 3–6 CrossRef PubMed; (b) W. L. Noorduin, W. J. P. Van Enckevort, H. Meekes, B. Kaptein, R. M. Kellogg, J. C. Tully, J. M. McBride and E. Vlieg, Angew. Chem., Int. Ed., 2010, 49, 8435–8438 CrossRef PubMed; (c) J. E. Hein, B. Huynh Cao, C. Viedma, R. M. Kellogg and D. G. Blackmond, J. Am. Chem. Soc., 2012, 134, 12629–12636 CrossRef PubMed; (d) M. Iggland and M. Mazzotti, Cryst. Growth Des., 2011, 11, 4611–4622 CrossRef; (e) C. Viedma, J. E. Ortiz, T. De Torres, T. Izumi and D. G. Blackmond, J. Am. Chem. Soc., 2008, 130, 15274–15275 CrossRef PubMed; (f) L. Spix, H. Meekes, R. H. Blaauw, W. J. P. Van Enckevort and E. Vlieg, Cryst. Growth Des., 2012, 12, 5796–5799 CrossRef; (g) B. Kaptein, W. L. Noorduin, H. Meekes, W. J. P. Van Enckevort, R. M. Kellogg and E. Vlieg, Angew. Chem., Int. Ed., 2008, 47, 7226–7229 CrossRef PubMed; (h) W. L. Noorduin, T. Izumi, A. Millemaggi, M. Leeman, H. Meekes, W. J. P. Van Enckevort, R. M. Kellogg, B. Kaptein, E. Vlieg and D. G. Blackmond, J. Am. Chem. Soc., 2008, 130, 1158–1159 CrossRef PubMed; (i) A. H. J. Engwerda, H. Meekes, B. Kaptein, F. P. J. T. Rutjes and E. Vlieg, Chem. Commun., 2016, 96, 1659–1676 Search PubMed; (j) W. L. Noorduin, B. Kaptein, H. Meekes, W. J. P. Van Enckevort, R. M. Kellogg and E. Vlieg, Angew. Chem., Int. Ed., 2009, 48, 4581–4583 CrossRef PubMed; (k) P. M. Björemark, S. Olsson, T. Kokoli and M. Håkansson, Chem. - Eur. J., 2015, 21, 8750–8753 CrossRef PubMed; (l) R. R. E. Steendam, J. M. M. Verkade, T. J. B. Van Benthem, H. Meekes, W. J. P. Van Enckevort, J. Raap, F. P. J. T. Rutjes and E. Vlieg, Nat. Commun., 2014, 5, 5543 CrossRef PubMed.
- (a) C. Rougeot, F. Guillen, J. C. Plaquevent and G. Coquerel, Cryst. Growth Des., 2015, 15, 2151–2155 CrossRef; (b) C. Xiouras, J. Van Aeken, J. Panis, J. H. Ter Horst, T. Van Gerven and G. D. Stefanidis, Cryst. Growth Des., 2015, 15, 5476–5484 CrossRef.
- C. Viedma and P. Cintas, Chem. Commun., 2011, 47, 12786 RSC.
- (a) K. Suwannasang, A. E. Flood, C. Rougeot and G. Coquerel, Cryst. Growth Des., 2013, 13, 3498–3504 CrossRef; (b) K. Suwannasang, A. E. Flood, C. Rougeot and G. Coquerel, Org. Process Res. Dev., 2017, 21, 623–630 CrossRef; (c) K. Suwannasang, A. E. Flood and G. Coquerel, Cryst. Growth Des., 2016, 16, 6461–6467 CrossRef; (d) W. W. Li, L. Spix, S. C. A. de Reus, H. Meekes, H. J. M. Kramer, E. Vlieg and J. H. ter Horst, Cryst. Growth Des., 2016, 6, 5563–5570 CrossRef; (e) F. Breveglieri, G. M. Maggioni and M. Mazzotti, Cryst. Growth Des., 2018, 18, 1873–1881 CrossRef; (f) R. Uchin, K. Suwannasang and A. E. Flood, Chem. Eng. Technol., 2017, 40, 1252–1260 CrossRef; (g) K. Suwannasang, G. Coquerel, C. Rougeot and A. E. Flood, Chem. Eng. Technol., 2014, 37, 1329–1339 CrossRef; (h) H. Katsuno and M. Uwaha, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2016, 93, 1–10 CrossRef PubMed.
- C. Xiouras, E. Van Cleemput, A. Kumpen, J. H. Ter Horst, T. Van Gerven and G. D. Stefanidis, Cryst. Growth Des., 2017, 17, 882–890 Search PubMed.
- C. R. Strauss and D. W. Rooney, Green Chem., 2010, 12, 1340 RSC.
- D. K. K. Kondepudi, R. J. J. Kaufman and N. Singh, Science, 1990, 250, 975–976 Search PubMed.
- The surface of the pre-existing crystals of each chirality acts as a catalyst for the formation of new nuclei of the same chirality (autocatalysis). Due to the fast racemization reaction, growing nuclei of both chiralities effectively compete for the same supersaturation (mutual inhibition).
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, solubility curves and crystal characterization. See DOI: 10.1039/c8ce00575c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |