
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A low-temperature synthesis method for AnO2 nanocrystals (An = Th, U, Np, and Pu) and associate solid solutions
Karin
Popa†
 *,
Olaf
Walter†
*,
Oliver Dieste
Blanco
,
Antony
Guiot
,
Daniel
Bouëxière
,
Jean-Yves
Colle
,
Laura
Martel
,
Mohamed
Naji‡
and
Dario
Manara
*,
Olaf
Walter†
*,
Oliver Dieste
Blanco
,
Antony
Guiot
,
Daniel
Bouëxière
,
Jean-Yves
Colle
,
Laura
Martel
,
Mohamed
Naji‡
and
Dario
Manara
European Commission, Joint Research Centre, P.O. Box 2340, D-76125 Karlsruhe, Germany. E-mail: karin.popa@ec.europa.eu; olaf.walter@ec.europa.eu
First published on 23rd July 2018
Abstract
Production of actinide oxide powder via dry thermal decomposition of corresponding oxalates is currently carried out on the industrial scale at temperatures exceeding 500 °C. Although it is simple, this method presents some disadvantages such as high decomposition temperature with a direct effect on the surface area, pre-organised morphology of the nanoparticles affecting the sintering behaviour, etc. We have recently proposed the decomposition of AnIV-oxalates under hot compressed water conditions as a straightforward way to produce reactive actinide oxide nanocrystals. This method could be easily applied at low temperatures (95–250 °C) in order to generate highly crystalline nano-AnO2. We present here the formation conditions of AnO2 (An = Th, U, Np, and Pu) and some associated solid solutions, their stability, and grain growth during thermal treatment. The involvement of water molecules in the mechanism of the oxalate decomposition under the hot compressed water conditions has been demonstrated by an isotopic exchange reaction during the thermal treatment of the hydrated oxalate in H2[17O] through MAS-NMR and Raman techniques.
1. Introduction
Oxalate precipitation is a versatile process often applied in the nuclear fuel cycle for different purposes (e.g. isotopic separation and preconcentration,1 quantitative recovery of actinide ions from waste solutions,2 fuel production,3 spent fuel treatment and reprocessing,4,5etc.). By precipitation with oxalic acid, the actinides form micrometre-sized hydrated oxalates, independent of the oxidation state of the actinide. Such oxalates can be converted to their corresponding oxides and gaseous products by thermal treatment, and thus the final products are free of impurities except eventually some residual carbon.6 Since the decomposition temperature is usually mild (500–800 °C), the final oxide is normally nanosized.7,8 However, the final powder consists of plate-like agglomerates, as a consequence of the pseudomorphic oxalate-to-oxide conversion. This morphology of the actinide oxalates may be altered by changing the basic precipitation conditions, as recently demonstrated in the thorium case.9 Another possibility would be to modify the decomposition conditions. For this purpose, we have recently proposed the conversion of the actinide oxalates under hot compressed water as an alternative to thermal decomposition of the oxalates. This method has been successfully applied to synthesize nanometric ThO2, UO2, and PuO2,10 and associate U1−xThxO2 solid solutions.11The attack of the oxalates under the hot compressed water conditions takes place between 95–250 °C, and thus, the AnO2 crystallites are even smaller than those in the conventional thermal route. This new route proved to be uncomplicated, fast, versatile, close to quantitative, and reproducible. It has fewer procedural steps than typical oxalate precipitation-thermal decomposition processes, thus allowing for production using a single vessel and under continuous flow. But the main achievement consists of the destructuring of the plate-like agglomerates specific to thermal conversion, with a direct effect on the sintering behaviour of the obtained nanomaterials.
With respect to other thermal methods employing organic solvents,12–14 the hydrothermal decomposition of oxalates presents the advantage that the material obtained is free of any residual carbon impurities possibly blocking the NCs surface.
In this paper, we extend our first report on the synthesis and characterisation of AnO2 (An = Th, U, and Pu) nanocrystals (NCs) formed by hydrothermal decomposition of An(C2O4)2·nH2O in batch reactors towards the actinide series Th, U, Np, and Pu, including synthetic trials for mixed oxide production. NpO2 NCs were characterised in detail by using spectroscopic and microscopic techniques. The influence of pressure on the crystallite size is also addressed. Moreover, the involvement of water molecules in the oxalate hydrothermal decomposition mechanism has been proven by studying the isotopic exchange reaction during the thermal decomposition of An(C2O4)2·nH2O in H2[17O] through MAS-NMR and Raman techniques. In order to assess the densification behaviour of AnO2 (An = Th, U, Np, and Pu) in more detail, we present here the growth of the particles as a function of temperature.
2. Experimental
2.1 Sample preparation
The decomposition of AnIV-oxalates under the hot compressed water conditions has been recently proposed by our group as a low-temperature method for the preparation of nano-AnO2. As starting materials, we have used ThIV (1.9 M in 8 M HNO3), UIV (0.47 M, obtained by electroreduction of a UO2(NO3)2 solution in 4 M HNO3 containing 0.5 M of hydrazine), NpIV (0.6 M in ca. 2 M HNO3), and PuIV (0.8 M in ca. 4 M HNO3). The other reagents (oxalic acid anhydrous, hydrazine hydrate, ceric ammonium nitrate, cerous nitrate hexahydrate, and H2[17O]) were of analytical grade and used as supplied by Merck or Sigma-Aldrich.
The oxalates An(C2O4)2·nH2O were firstly produced by direct precipitation of the AnIV solution in nitric acid with a stoichiometric amount of the oxalic acid 1 M solution.15–19 A similar method was applied for different mixed oxalates. A possible alternative approach for the production of mixed oxides is to start from oxalate end-members. The obtained precipitates were washed several times with distilled water in order to remove any trace of nitrate, which induces the oxidation of UIV to soluble UVI under the working temperature conditions.
Most of the experimental trials were performed in 25 ml Teflon-lined hydrothermal synthesis autoclave reactors. The heating stages were performed by using a heating mantel preheated at the requested temperature while the temperature was controlled using thermocouples. As compared to classical thermal oxalate decomposition, this method employs not only temperature, but also autogenic pressure to achieve the conversion into the oxide. For this purpose, small amounts of water were added to the powder. Hence, inside the autoclave, the increasing temperature caused the formation of water vapour and other gases which raised the pressure in the vessel. The following general reaction occurs (eventually with dehydration steps):
| An(C2O4)2·nH2O → AnO2 + 2CO + 2CO2 + nH2O | (1) |
For the Pu-containing compositions, the formation of the PuIII-oxalate intermediate might be possible.20,21 Finally, the resulting NCs were washed with water, ethanol, and acetone, in order to gradually decrease the polarity of the solution. In the case of the uranium-containing samples, the work was performed under argon and using minute amounts of hydrazine, in order to limit the oxidation of UIV.
The influence of the pressure on the NC sizes of ThO2 and UO2(+x) was studied by using 25 ml Paar high pressure/high temperature vessels made out of stainless steel. Thus, 25 mg of AnIV-oxalate was decomposed in the presence of different volumes of water (1–10 ml) for 30 min at 400 ± 15 °C (preheated system) without stirring.
Nano-CeO2 used as a reference for the study of the thermal behaviour was obtained by thermal decomposition of Ce-oxalate.
| Th(C2[16O]4)2·2H2[16O] + H2[17O] → Th[16,17O]2 + 2C[16,17O] + 2C[16,17O]2 + 3H2[16,17O] | (2) |
Thorium has been chosen in this experiment because of its resistance to oxidation (in contrast to nano-UO2 and associated solid solutions, nano-ThO2 does not oxidise as a function of time) and its low radioactivity. Thus, parallel experiments were performed by decomposing 50 mg of Th(C2O4)2·2H2O in the presence of 0.5 ml H2[16O] or H2[17O] for 18 h at 250 °C.
2.2 Physicochemical characterisation
A Philips XL40 scanning electron microscope (SEM) equipped with an energy dispersive X-ray spectroscope (EDS) was used. The sample grains were deposited on the usual carbon sticker and covered with carbon to avoid charging.
Infrared measurements were performed on the solid samples using a Bruker Alpha Platinum spectrometer using a horizontal ZnSe ATR (attenuated total reflection) crystal.
The 17O MAS NMR spectra were acquired on a Bruker 9.4 T at a Larmor frequency of 54.25 MHz.24 As 17O is a quadrupolar nucleus (I = 5/2), a very short pulse25 of 1 μs was used to acquire the spectrum using a single pulse experiment. The spectrum was referenced at 0 ppm to liquid H217O.
3. Results and discussion
3.1 Synthesis of AnO2 (An = U, Th, Np, and Pu) and associated solid solutions through the decomposition of the oxalates under the hot compressed water conditions
As can be seen from the XRD data shown in Fig. 1, well defined NCs (fluorite structure, cubic Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (225) space group) are formed in all cases which is reflected by the appearance of broad peaks at up to high angles in the diffractograms. No significant weight loss potentially associated with the solvent adsorbed onto the NCs surface has been detected.
m (225) space group) are formed in all cases which is reflected by the appearance of broad peaks at up to high angles in the diffractograms. No significant weight loss potentially associated with the solvent adsorbed onto the NCs surface has been detected.
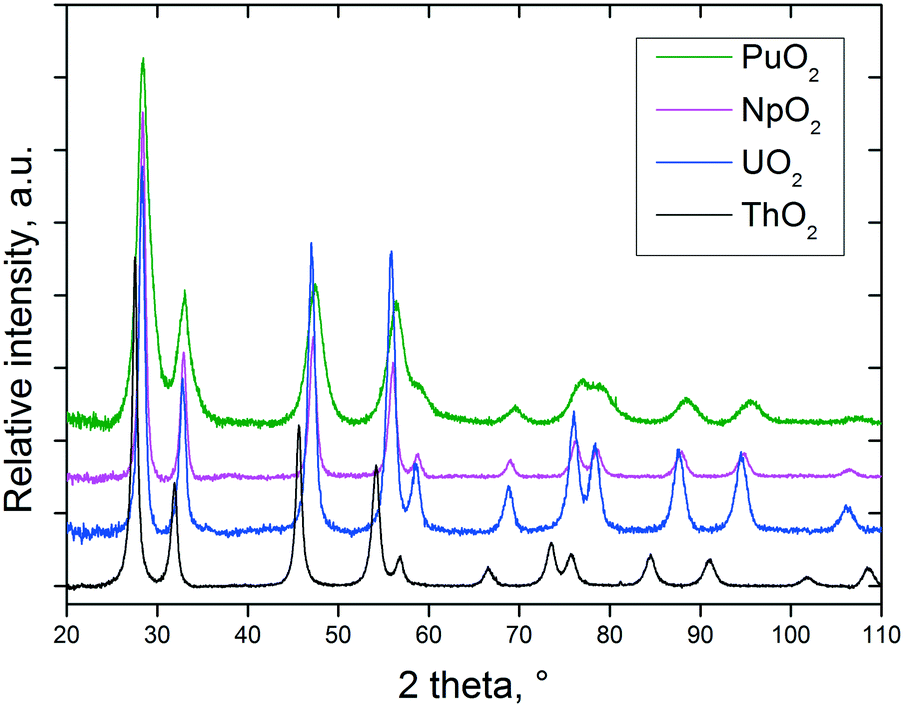 | ||
| Fig. 1 XRD patterns of nanocrystalline AnO2 (An = Th, U, Np, and Pu) obtained by decomposition of the corresponding oxalates under the hot compressed water conditions. | ||
The particle sizes were calculated from the XRD data based on the full width at half maximum of at least six selected peaks in the 2θ range between 25 and 100°. In some cases, the crystallite sizes were also assessed by direct TEM observation (Table 1). The results obtained from the two characterisation techniques are usually in good agreement.
| Compound | Reaction conditions used for An(C2O4)2·nH2O decomposition | a lattice parameter, Å | Particle size, nm | Colour |
|---|---|---|---|---|
| ThO2 | 3.5 h/250 °C, 3 ml water, 2000 rpm | 5.611(1) | 6.1 ± 0.7 (XRD)/5.5 ± 0.9 (TEM) | White |
| ThO2 | 3 h/250 °C, 5 ml water, 2000 rpm | 5.610(1) | 6.8 ± 1.1 (XRD) | White |
| ThO2 | 18 h/250 °C, 4 ml water, 2000 rpm | 5.613(1) | 7.1 ± 0.9 (XRD)/6.6 ± 1.2 (TEM) | White |
| UO2(+x) | 18 h/250 °C, 20 μl (NH2)2, 3 ml water, 2000 rpm | 5.454(2) | 5.5 ± 0.5 (XRD)/5.2 ± 0.6 (TEM) | Black |
| UO2(+x) | 4 h/250 °C, 100 μl (NH2)2, 5 ml water, 2000 rpm | 5.461(2) | 6.3 ± 0.6 (XRD)/5.9 ± 0.7 (TEM) | Black |
| UO2(+x) | 3 h/250 °C, 50 μl (NH2)2, 5 ml water, 2000 rpm | 5.467(2) | 7.2 ± 0.8 (XRD) | Black |
| UO2(+x) | 3.5 h/170 °C, 10 μl (NH2)2, 3 ml water, 2000 rpm | 5.463(2) | 7.8 ± 0.9 (XRD) | Black |
| UO2(+x) | 4 h/170 °C, 1000 μl (NH2)2, 7 ml water, 25 bar, no stirring (Paar autoclave) | 5.459(2) | 4.5 ± 0.5 (XRD) | Black |
| PuO2 | 120 h/95 °C, 5 ml water, no stirring | 5.397(1) | 3.7 ± 1.0 (XRD)/2.6 ± 0.5 (TEM) | Green |
| NpO2 | 18 h/160 °C, 3 ml water, no stirring | 5.433(1) | 9.6 ± 1.8 (XRD) | Beige |
| NpO2 | 18 h/200 °C, 5 ml water, no stirring | 5.441(1) | 7.5 ± 1.6 (TEM) | Beige |
| Th0.2U0.8O2 | 4 h/170 °C, 70 μl (NH2)2, 3 ml water, 2000 rpm | 5.504(2) | 3.8 ± 0.5 (XRD) | Black |
| Th0.5U0.5O2 | 18 h/250 °C, 15 μl (NH2)2, 4 ml water, 2000 rpm | 5.542(2) | 6.4 ± 1.1 (XRD) | Grey |
| Th0.5U0.5O2 | 18 h/250 °C, 20 μl (NH2)2, 2 ml water, 2000 rpm | 5.541(1) | 3.1 ± 0.4 (XRD) | Grey |
| Th0.44Pu0.56O2 | Mixture of the two end members, 70 h/170 °C, no stirring | 5.481(4) | 4.4 ± 0.9 (XRD) | Greenish |
| Np0.33Pu0.67O2 | 18 h/200 °C, 5 ml water, no stirring | 5.411(1) | 8.4 ± 1.6 (XRD)/6.6 ± 2.5 (TEM) | Light grey |
| Th0.70U0.15Np0.15O2 | 18 h/200 °C, 5 ml water, no stirring | 5.530(1) | 7.0 ± 2.5 (XRD)/6.7 ± 1.1 (TEM) | Brown |
| Th0.60Np0.20Pu0.20O2 | 18 h/200 °C, 5 ml water, no stirring | 5.520(2) | 5.0 ± 1.1 (XRD)/5.1 ± 1.4 (TEM) | Light green |
The temperature of the particle formation increases from Pu to Th, i.e. (i) ThO2 NCs can only be obtained at reaction temperatures of 250 °C; (ii) UO2 NCs are nicely formed at temperatures above 150 °C; (iii) for the formation of the NpO2 NCs, a temperature of 160–200 °C was applied (the reaction temperature is not yet optimised); and (iv) the PuO2 NCs are already formed at 95 °C after several days.
Due to the low-temperature decomposition, the size of the PuO2 NCs is smallest, which is obvious from the increased line broadening observed in Fig. 1. In the case of the uranium dioxide NCs, it turned out that it is of real advantage to work in anoxic conditions, as otherwise (especially at low temperatures and long reaction times), even complete oxidation of all the UIV to UVI might proceed; UO2(OH)2 was found at the bottom of the yellow solutions as the crystalline product in some experiments. No sign of oxidation of uranium has been detected in the freshly prepared urania samples, additionally confirmed by the absence of the characteristic IR band at 740 cm−1 specific to the stretching vibrations of the –U–O–U–O– chain.26 Furthermore, it is essential, in order to control the size of the uranium dioxide NCs, to work in a non-oxidising environment.
Here, one has to note the divergent decomposition behaviour of the plutonium and cerium oxalates. In both cases, it was reported that the reduction of the tetravalent ion to the trivalent one occurs during the oxalate precipitation.27,28 Our results indicate that the decomposition of the plutonium oxalate hexahydrate under the hot compressed water conditions leads to the formation of PuO2 NCs. At least in this particular case (reaction conducted at 95 °C), transformation occurs with no evidence of change in the oxidation state since we do not reach temperatures of 150–220 °C, potentially needed for the reduction of PuIV- to PuIII-oxalate in the presence of carbon monoxide.29 On the other hand, the product of the cerium oxalate decomposition at 400 °C/250 bar is hexagonal Ce(CO3)(OH), in which cerium has a trivalent oxidation state. Ce(CO3)(OH) is isostructural with the hydroxylbastnäsite-(Ce) mineral30 and several other lanthanide hydroxocarbonates.31
The TEM analysis indicates that the typical spherical agglomerates of mixed oxides resulted from the decomposition of the mixed oxalates with about 100–200 nm in size, reaching 600 nm in some rare cases, which in general do not keep the plate-like morphology specific to the pseudomorphic thermal conversion of oxalate (Fig. 2). In a previous work (Balice et al.),11 we potentially attributed the morphology of the agglomerates (i) to the stirring applied during the decomposition process under the hot compressed water conditions or (ii) to a different reaction mechanism while proceeding in water. The present results clearly indicate that similar shaped reaction products are obtained independent of the stirring process (Table 1); thus, the first hypothesis may be excluded at this point.
 | ||
| Fig. 2 Increasing magnification images on typical aggregates of NCs as seen on every sample. | ||
A NpO2 sample has been extensively characterised by TEM. The size of the observed crystallites has been statistically studied, obtaining an average value of 7.5 ± 1.6 nm. In the same manner as the other nanoparticles already studied, the crystallites are not colloidal, but agglomerated into quasi-spherical groups with sizes of 0.3(1) μm. These aggregates consist of crystallites with low relative angle orientations, as shown in Fig. 3. Some of these agglomerations present quasi-monocrystalline diffraction, as shown in the high resolution micrograph, which is an indication of the fact that the process of nano-restructuring has not been completed, and the newly created crystallites remain attached to their original positions. For the case of NpO2, the expected oxidation state is IV, corresponding to an f-occupancy of 3; the EELS calculations done on the collected spectra showed an average oxidation of +3.73 ± 0.50 (f-occupancy of 3.27), which is slightly lower than expected; due to the nanocrystalline nature of the sample, it is likely that variations on the oxidation state between the bulk and the surface of the sample are to be found. For this reason, the oxidation state obtained may vary depending on where exactly the measurement was performed.
 | ||
| Fig. 3 (a) Different agglomerates showing small angle relative orientations of the NpO2 crystallites within them. (b) High resolution micrograph proving the nano-restructuring of a larger single crystal. | ||
For the solid solutions we report here, the lattice parameter agrees fairly with the expected theoretical values (Table 1). In the special case of several compositions containing thorium (Th–Np and Th–U–Pu), a mixture of two cubic phases has been obtained, because of the reduced reactivity of thorium.32,33
The studied mixed oxide crystallites do not arrange randomly on these agglomerates, but rather maintain part of their former orientation in the same manner as what happened on the NpO2 sample, as proved by the non-uniformly distributed diffraction spots around the centre of the electron diffraction patterns (Fig. 4a): the electron diffraction of the crystallites on each agglomerate form a small angle with each other, in the same manner as observed for NpO2, indicating again an incomplete nano-restructuring process. The elemental quantification performed on each sample by means of the second derivative method on the collected EELS spectra showed no difference in composition between all the studied agglomerates (Fig. 4b).
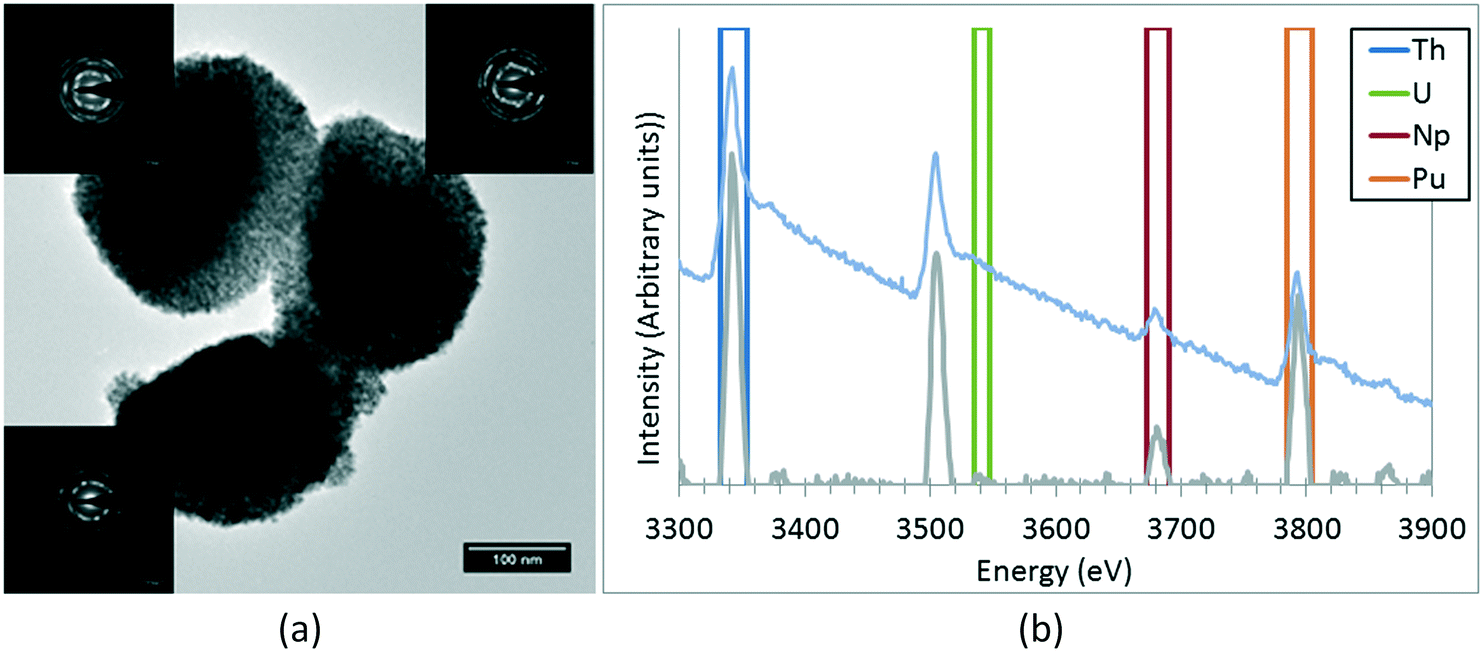 | ||
| Fig. 4 (a) Details on the low-angle alignment of the crystallites on each aggregate observed on every sample. Each electron diffraction pattern has been obtained exclusively from the adjacent aggregate. (b) Example of an EELS spectrum with the 2nd derivative and the windows used for the integration of each peak. | ||
Raman spectroscopy measurements performed on the Np0.33Pu0.67O2, Th0.70U0.15Np0.15O2, and Th0.60Np0.20Pu0.20O2 compositions confirm the formation of a fluorite-structured mixed dioxide. This can be promptly deduced from the observation of a dominant T2g Raman mode in the spectral region between 460 cm−1 and 480 cm−1.34 It is seen in Fig. 5 that all spectra present asymmetry at the low energy part of the T2g band for selected compositions, in agreement with earlier work.35 In order to estimate the ThO2 crystal size, we fitted the T2g line to a Lorenzian band and projected the obtained full-width at half maximum value in the FWHM vs. crystal size curve already obtained on ThO2 by Cappia's work.35 Accordingly, for a FWHM (T2g) = 11 ± 2 cm−1, a crystal size of L = 10–18 nm is calculated, not in fair agreement with the data obtained from XRD and TEM (Table 1). The discrepancy might arise mainly from the considered spherical dispersion of (LO1, TO1) and the fact that their splitting over the Brillouin zone is neglected in the present analysis.
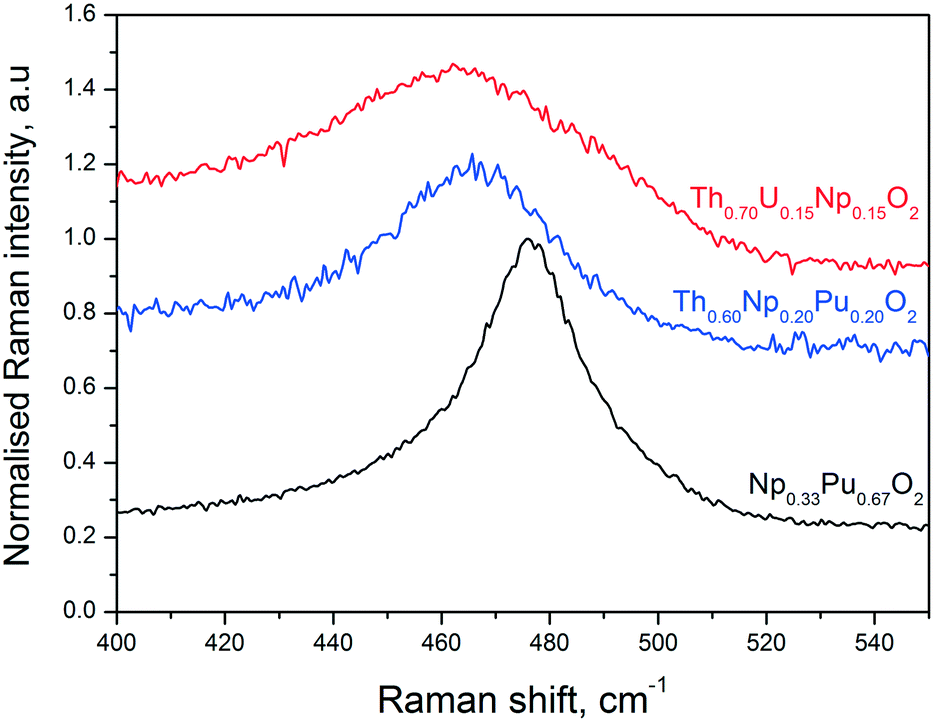 | ||
| Fig. 5 Example Raman spectra recorded on three of the present samples, in the T2g line spectral region, using a laser wavelength of 647 cm−1. | ||
The position of the T2g band in the Raman spectra of the mixed oxide specimens varies slightly but consistently depending on the sample composition (Fig. 5). As compared to the pure actinide dioxides, in which the T2g spectral positions are well established (e.g. 464 cm−1 for ThO2, 467 cm−1 for NpO2, and 478 cm−1 for PuO2),36 a decrease of the T2g intensity and band broadening is observed for the mixed solid solutions in the Raman spectra. The observation that the T2g band of the mixed solid solutions has its maximum at an intermediate composition results from the convolution of the single metal-oxygen bond vibrations. More precisely, one can resolve two absorptions, by fitting the mixed oxide Raman spectra, attributed to the presence of two-mode behaviour, where each single mode arises from one specific anion-cation vibration.37 Independently, the current T2g peaks signify the presence of mixed solid solutions with compositions in reasonable agreement to the ones obtained by other analytical methods.
3.2 The role of water in the decomposition of the oxalates under the hot compressed water conditions
In order to explore the role of water more in detail, we performed an experiment for the thermal decomposition of Th(C2O4)2·2H2O in more than 90% enriched H2[17O] and then analysed the product via Raman spectroscopy and compared it with the product obtained in normal water. Fig. 6 shows the Raman spectra of the two forms of thorium dioxide (Th[16O]2 and Th[17O]2) recorded with an excitation energy of Ei = 1.91 eV (647 nm). Up to 1200 cm−1, the spectra are dominated by the T2g ∼ 460 cm−1 Raman feature for the spectrum of fluorite.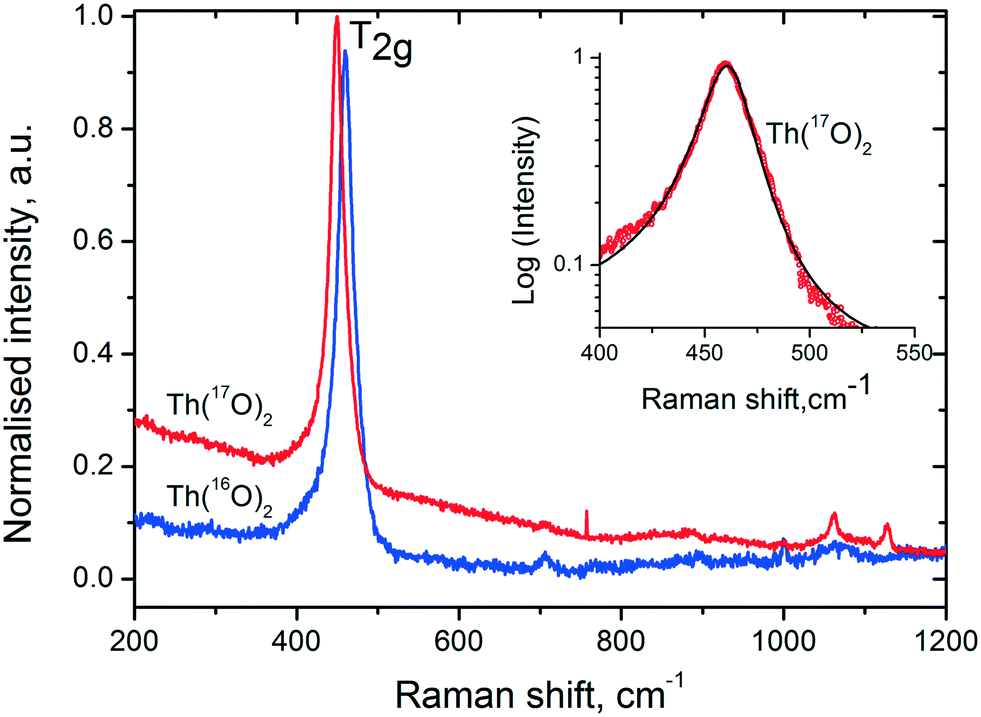 | ||
| Fig. 6 Raman spectra of Th[16O]2 and Th[17O]2. Insert shows the asymmetric nature of the T2g mode (red symbols) fitted together with the asymmetric Lorentzian function. | ||
In addition to the phonon confinement feature (discussed above), the T2g mode of the Raman spectrum of an 17O enriched sample shows a downshift of ∼11 cm−1, as compared to that of 16O. If we consider the one phonon excitation, the vibration frequency is expected to vary proportionally to the square root of the mass:
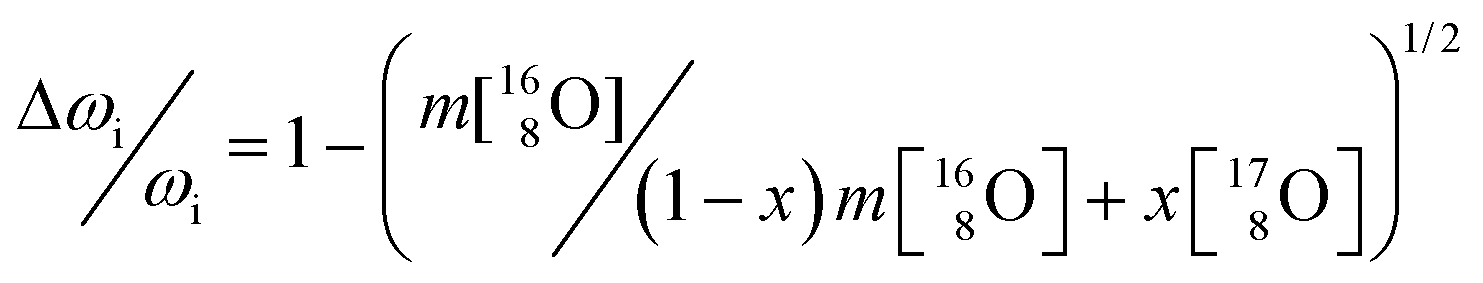 | (3) |
As 17O is the only NMR active oxygen isotope, this technique is very appropriate and complementary to Raman spectroscopy in determining whether the 17O coming from water has been incorporated in the ThO2 matrix. There is good agreement (Fig. 7) between the 17O MAS NMR spectra of crystalline (bulk) ThO2 (main peak at 576 ppm) and of nano-ThO2 (main peak at 575 ppm). Thus, the samples possess very similar chemical shifts underlining a similar local environment, but the full width at half maximum (FWHM) of the main peak increases from 3 to 7 ppm, respectively. This FWHM increase is the spectral print of the nanomaterials as observed previously.38 This further supports the results discussed above.
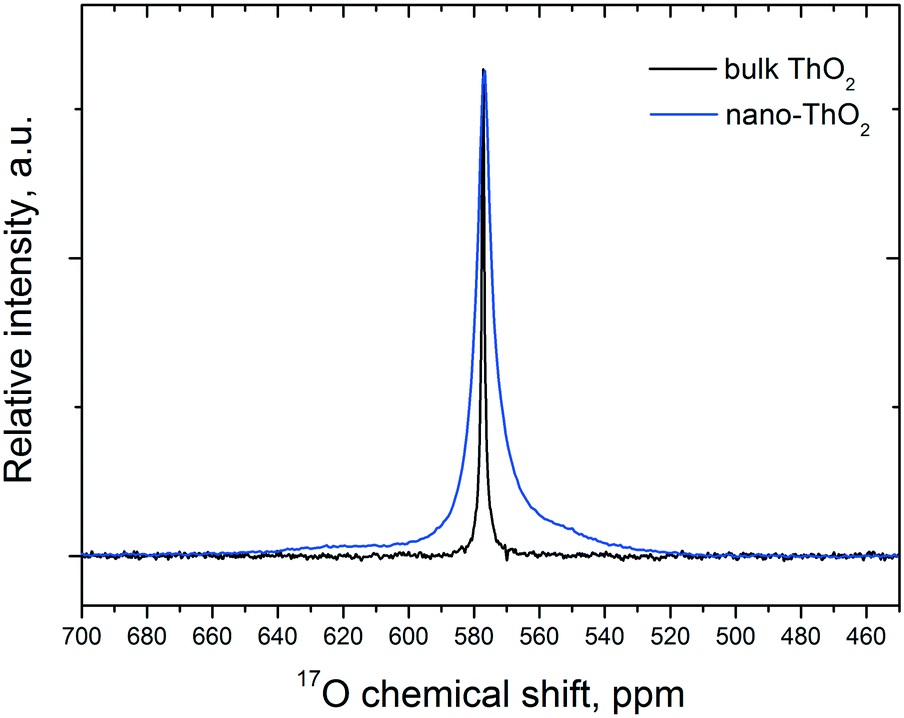 | ||
| Fig. 7 17O MAS NMR spectra of the bulk and nano-Th[17O]2 acquired at 50 kHz. | ||
3.3 The thermal behaviour of the AnO2 NCs
NCs offer the possibility to study the sintering behaviour of AnO2 in more detail, e.g. the growth of the particles as a function of temperature or the temperature dependence of the lattice parameters (Fig. 8). This examination contributes to the knowledge on how fast particles can change their shape in nuclear fuel in which temperatures up to 1500 °C are easily reached in the centre of the fuel. In particular, when the particles are small, significant growth can be detected already at much lower temperatures.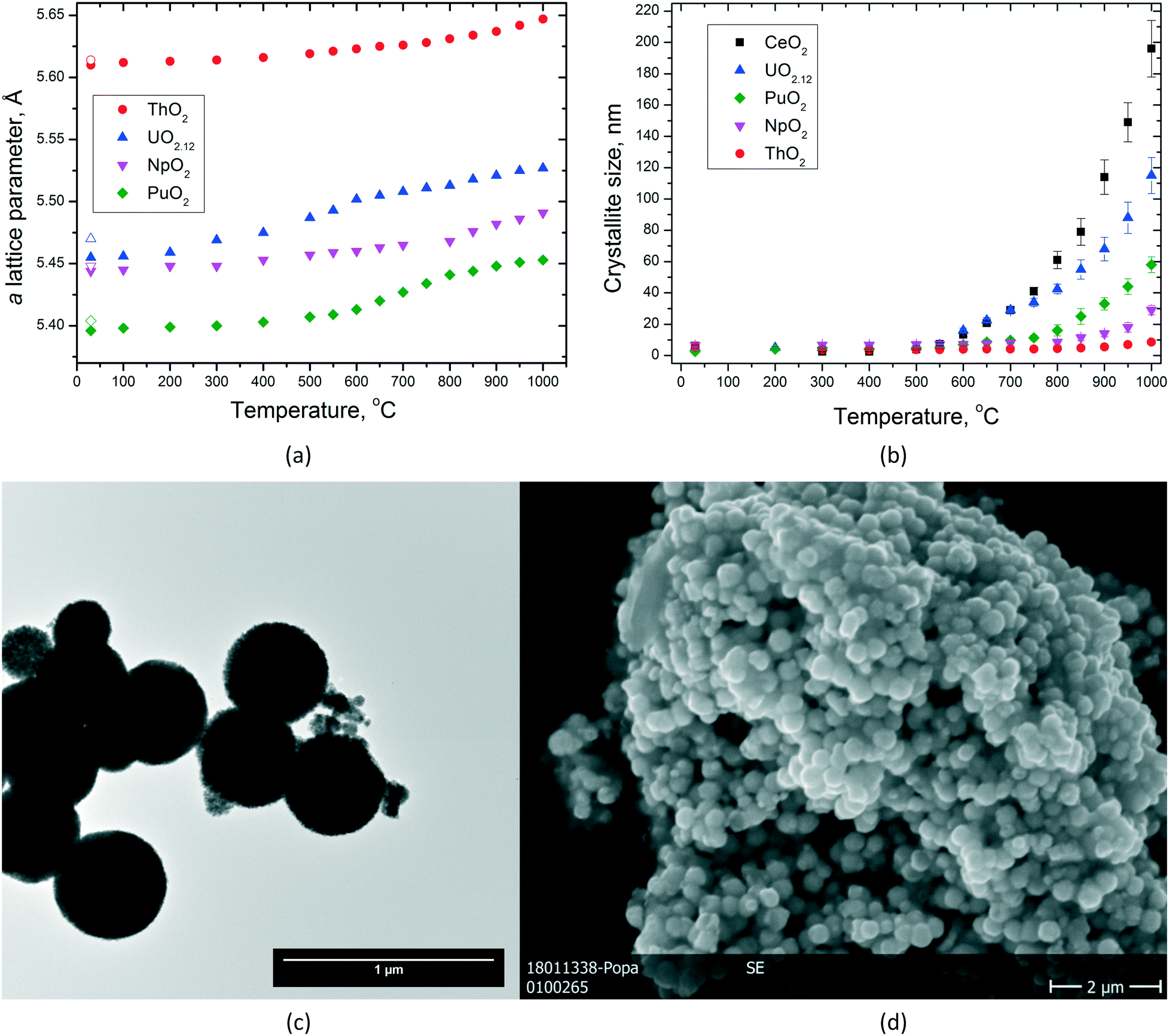 | ||
| Fig. 8 Variation of the lattice parameters (empty symbols indicate the values after cooling) (a) and of the particle size (b) of AnO2 NCs as a function of temperature. (c) TEM images of the original NpO2 used in this experiment, indicating the size of the agglomerates. (d) SEM image of the same NpO2 specimen after heating at 1000 °C; note that the grain size fits well with the dimensions of the agglomerates in the original powder. | ||
The very limited crystallite growth of ThO2 below 1000 °C compared to UO2(+x) (ref. 39) is in agreement with the lower self-diffusion of ThIV already discussed in our previous paper (higher melting point and higher sintering temperature).11 The initial urania powder used during this experiment was hyperstoichiometric (UO2.12), so several processes are possible during heating till 800 °C, until the powder turned into fully stoichiometric UO2.00. The grain growth of NpO2 NCs is also very limited in the studied temperature range, with a direct effect in the formation of monophasic Np-containing mixed oxides. However, it can be seen that all the dioxides show particle growth by increasing temperature, but the obtained particle size for each given temperature differs from one oxide to the other (Fig. 8b). The largest particles are obtained for CeO2 followed by UO2, PuO2, NpO2, and ThO2, which showed only very little particle growth.
By analysing the graph for the lattice parameters as a function of temperature (Fig. 8a), one can see that of course thermal expansion takes place in all cases leading to a constant increase of the cell parameter a. This is well reflected in the curve obtained for ThO2. However, the shapes of the curves are not identical. Whereas for ThO2, one can see a constant increase in the lattice parameter a; for UO2, PuO2, and NpO2, two different parts in the curves can be identified like there would be a step in between. This step is observed at different temperatures, it can be assigned to UO2 at ca. 600 °C, to PuO2 at ca. 750 °C, and to NpO2 at ca. 850 °C. After this jump, the growth proceeds linearly due to thermal expansion like before (nicely seen for UO2 and PuO2, Fig. 8a). This observation is somehow in agreement with the temperature at which particle growth is started to be observed; therefore, we correlate it to a kind of sintering of very small crystallites with still surface-controlled properties (Fig. 8d). At higher temperatures, the crystal lattice parameter does not show any surface dependency anymore and the materials behave more like the bulk.
3.4 Influence of the pressure on the crystallite size
Besides the effect of temperature, the dimensions of the NCs can be influenced as well by pressure. Accordingly, we studied, for example, the size of the particles of UO2 and ThO2 formed during the synthesis as a function of pressure at a given temperature of 400 °C. However, only on a very limited range between ambient pressure and close to 300 bar could be studied due to the limitations of the reactor (Fig. 9).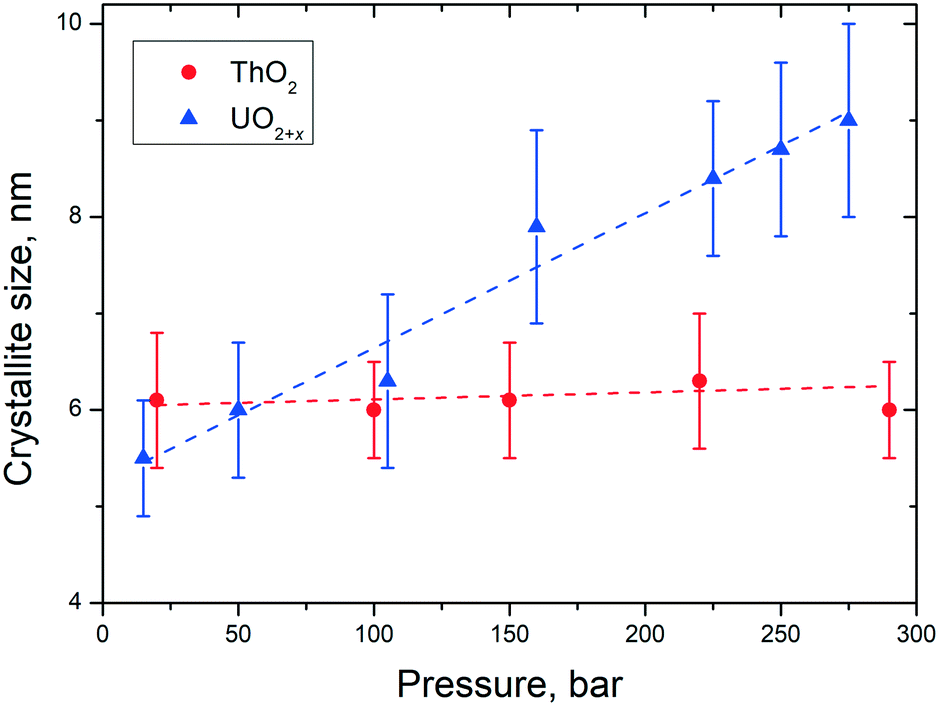 | ||
| Fig. 9 Variation of the particle size of AnO2 NCs as a function of pressure at a temperature of 400 ± 15 °C. | ||
In the pressure range of up to 300 bar, no pressure effect on the size of the ThO2 particles can be observed. In contrast, the particles size of urania increased from 5.5 nm (10 bar) to 9 nm (250 bar). These findings are in agreement with the ones described before for the temperature influence on the particles size. UO2(+x) NCs agglomerate more easily than the ThO2 ones which correlates to their sintering properties.
4. Conclusions and perspectives
We present here an effective and easy way of producing small NCs of AnO2 (size typically less than 10 nm) on the laboratory scale by decomposition of An(C2O4)2·nH2O under hot compressed water conditions at temperatures usually lower than 250 °C. This method presents several advantages compared with the thermal conversion of oxalates (lower particle size and avoiding the plate-like morphology) or the (mild) hydrothermal attack on other actinide compounds in non-aqueous solutions (the graphite layer on the surface of the NCs).This approach exhibits a key step forward towards the production of sinterable nanosized AnO2 powders (pure end members and mixed oxides) since the process exhibits potential for scale-up. This method could be potentially used for the incorporation of tetravalent americium in nanocrystalline mixed oxides. This method is versatile and can easily be applied for the synthesis of mixed oxides which are accessible by co-precipitation of the oxalate followed by treatment under hot compressed water afterwards. A possible alternative approach is to start from oxalate end-members. However, this is yet to be evaluated more in detail.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Joe Somers and Rudy Konings for their support and fruitful discussions. The technical support of Luca Balice, Bert Cremer, Herwin Hein and Sarah Stohr is highly appreciated. This work contributes to the Joint Programme on Nuclear Materials (JPNM) of the European Energy Research Alliance (EERA).References
- N. Vaida and C.-K. Kim, J. Radioanal. Nucl. Chem., 2010, 284, 341–366 CrossRef.
- N. Xu, D. Gallimore, E. Lujan, K. Garduno, L. Walker, F. Taylor, P. Thompson and L. Tandon, J. Radioanal. Nucl. Chem., 2016, 307, 1203–1213 CrossRef.
- B. Arab-Chapelet, P. M. Martin, S. Costenoble, T. Delahaye, A. C. Scheinost, S. Grandjean and F. Abraham, Dalton Trans., 2016, 45, 6909–6919 RSC.
- R. Taylor, Reprocessing and recycling of spent nuclear fuel, Woodhead Publishing, 2015 Search PubMed.
- P. Govindan, A. Palamalai, K. S. Vijayan, M. Raja, S. Parthasarathy, S. V. Mohan and R. V. Subba Rao, J. Radioanal. Nucl. Chem., 2000, 246, 441–444 CrossRef.
- Y. Ziouane, B. Arab-Chapelet, C. Tamain, S. Lalleman, T. Delahaye and G. Leturcq, J. Solid State Chem., 2016, 244, 45–51 CrossRef.
- V. Tyrpekl, J.-F. Vigier, D. Manara, T. Wiss, O. Dieste Blanco and J. Somers, J. Nucl. Mater., 2015, 460, 200–208 CrossRef.
- V. Tyrpekl, M. Cologna, D. Robba and J. Somers, J. Eur. Ceram. Soc., 2016, 36, 767–772 CrossRef.
- V. Tyrpekl, M. Beliš, T. Wangle, J. Vleugels and M. Verwerft, J. Nucl. Mater., 2017, 493, 255–263 CrossRef.
- O. Walter, K. Popa and O. Dieste Blanco, Open Chem., 2016, 14, 170–174 Search PubMed.
- L. Balice, D. Bouëxière, M. Cologna, A. Cambriani, J.-F. Vigier, E. De Bona, D. G. Sorarù, C. Kübel, O. Walter and K. Popa, J. Nucl. Mater., 2018, 498, 307–313 CrossRef.
- Q. Wang, G.-D. Li, S. Xu, J.-X. Li and J.-S. Chen, J. Mater. Chem., 2008, 18, 1146–1152 RSC.
- D. Hudry, C. Apostolidis, O. Walter, A. Janβen, D. Manara, J.-C. Griveau, E. Colineau, T. Vitova, T. Prüβmann, D. Wang, C. Kübel and D. Meyer, Chem. – Eur. J., 2014, 20, 10431–10438 CrossRef PubMed.
- R. Zao, L. Wang, Z.-F. Chai and W.-Q. Shi, RSC Adv., 2014, 4, 52209–52214 RSC.
- M. S. Girogor'ev, I. A. Charushnikova, N. N. Krot, A. I. Yanovskii and Y. T. Struchkov, Radiochemistry, 1997, 39, 420–423 Search PubMed.
- N. Clavier, N. Hingant, M. Rivenet, S. Obbade, N. Dacheux, N. Barré and F. Abraham, Inorg. Chem., 2010, 49, 1921–1931 CrossRef PubMed.
- F. Abraham, B. Arab-Chapelet, M. Rivenet, C. Tamain and S. Grandjean, Coord. Chem. Rev., 2014, 266–267, 28–68 CrossRef.
- C. Tamain, B. Arab-Chapelet, M. Rivenet, X. F. Legoff, G. Loubert, S. Grandjean and F. Abraham, Inorg. Chem., 2016, 55, 51–61 CrossRef PubMed.
- C. Tamain, B. Arab-Chapelet, M. Rivenet, S. Grandjean and F. Abraham, J. Solid State Chem., 2016, 236, 246–256 CrossRef.
- L. De Almeida, S. Grandjean, N. Vigier and F. Pattison, Eur. J. Inorg. Chem., 2012, 4986–4999 CrossRef.
- R. M. Orr and H. E. Sims, J. Nucl. Mater., 2015, 465, 756–773 CrossRef.
- G. van der Laan, K. T. Moore, J. G. Tobin, B. W. Chung, M. A. Wall and A. J. Schwartz, Phys. Rev. Lett., 2004, 93, 094401 CrossRef PubMed.
- V. J. Keast, Mater. Charact., 2012, 73, 1–7 CrossRef.
- L. Martel, N. Magnani, J.-F. Vigier, J. Boshoven, C. Selfslag, I. Farnan, J.-C. Griveau, J. Somers and T. Fanghänel, Inorg. Chem., 2014, 53, 6928–6933 CrossRef PubMed.
- D. Massiot, C. Bessada, J. P. Coutures and F. Taulelle, J. Magn. Reson., 1990, 90, 231–242 Search PubMed.
- G. C. Allen and N. R. Holmes, Appl. Spectrosc., 1994, 48, 525–530 CrossRef.
- I. L. Jenkins and M. J. Waterman, J. Inorg. Nucl. Chem., 1964, 26, 131–137 CrossRef.
- A. I. Karelin, N. N. Krot, R. D. Koslova, V. A. Lobas and V. A. Matukha, J. Radioanal. Nucl. Chem., 1990, 143, 241–252 CrossRef.
- N. Vigier, S. Grandjean, B. Arab-Chapelet and F. Abraham, J. Alloys Compd., 2007, 444–445, 594–597 CrossRef.
- K. Michiba, R. Miyawaki, T. Minakawa, Y. Terada, I. Nakai and S. Mathubara, J. Mineral. Petrol. Sci., 2013, 108, 326–334 CrossRef.
- K. Michiba, T. Tahara, I. Nakai, R. Miyawaki and S. Mathubara, Z. Kristallogr., 2011, 226, 518–530 Search PubMed.
- G. I. Nkou Bouala, N. Clavier, J. Léchelle, J. Monnier, C. Ricolleau, N. Dacheux and R. Podor, J. Eur. Ceram. Soc., 2017, 37, 727–738 CrossRef.
- N. Clavier, G. I. Nkou Bouala, J. Léchelle, J. Martinez, N. Dacheux and R. Podor, Radiochim. Acta, 2017, 105, 879–892 Search PubMed.
- V. G. Keramidas and W. B. White, J. Chem. Phys., 1973, 59, 1561–1562 CrossRef.
- F. Cappia, D. Hudry, E. Courtois, A. Janβen, L. Luzzi, R. J. M. Konings and D. Manara, Mater. Res. Express, 2014, 1, 025034 CrossRef.
- G. Begun, R. Haire, W. Wilmarth and J. Peterson, J. Less-Common Met., 1990, 162, 129–133 CrossRef.
- R. Rao, R. K. Bhagat, N. P. Salke and A. Kumar, Appl. Spectrosc., 2014, 68, 44–48 CrossRef PubMed.
- V. Sepelak, J. Mater. Chem., 2011, 21, 8332–8337 RSC.
- R. V. Krishnan, G. Panneerselvam, P. Manikandan, M. P. Antony and K. Nagarajan, J. Nucl. Radiochem. Sci., 2009, 10, 19–26 Search PubMed.
Footnotes |
| † These authors contributed equally to this work. |
| ‡ Current address: University Sidi Mohamed ben Abdellah, Department of Physics -Dhar El Mehraz, B.P. 1796 Atlas Fez, Morocco. |
| This journal is © The Royal Society of Chemistry 2018 |