
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning charge-assisted and weak hydrogen bonds in molecular complexes of the proton sponge DMAN by acid co-former substitution†
Lucy K.
Saunders
 *a,
Harriott
Nowell
a,
Helen C. E.
Spencer
b,
Lauren E.
Hatcher
b,
Helena J.
Shepherd
bc,
Lynne H.
Thomas
b,
Charlotte L.
Jones
b,
Simon J.
Teat
d,
Paul R.
Raithby
be and
Chick C.
Wilson
b
*a,
Harriott
Nowell
a,
Helen C. E.
Spencer
b,
Lauren E.
Hatcher
b,
Helena J.
Shepherd
bc,
Lynne H.
Thomas
b,
Charlotte L.
Jones
b,
Simon J.
Teat
d,
Paul R.
Raithby
be and
Chick C.
Wilson
b
aDiamond Light Source, Harwell Science and Innovation Campus, Didcot, Oxfordshire OX11 0DE, UK. E-mail: lucy.saunders@diamond.ac.uk
bDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK
cSchool of Physical Sciences, University of Kent, Canterbury, Kent CT2 7NH, UK
dAdvanced Light Source, Lawrence Berkeley National Lab, Berkeley, CA 94270, USA
eResearch Complex at Harwell, Harwell Science and Innovation Campus, Didcot, Oxfordshire OX11 0DE, UK
First published on 9th May 2018
Abstract
Nine new molecular complexes of the proton sponge 1,8-bis(dimethylamino)naphthalene (DMAN) with substituted benzoic acid co-formers have been engineered with varying component stoichiometries (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:2 or 1:3). These complexes are all ionic in nature, following proton transfer between the acid co-former and DMAN; the extracted proton is held by DMAN in all instances in an intramolecular [N–H⋯N]+ hydrogen bond. A number of structural features are common to all complexes and are found to be tunable in a predictable way using systematic acid co-former substitution. These features include charge-assisted hydrogen bonds formed between acid co-formers in hydrogen bonding motifs consistent with complex stoichiometry, and weak hydrogen bonds which facilitate the crystal packing of DMAN and acid co-former components into a regular motif. Possible crystal structure tuning by co-former substitution can aid the rational design of such materials, offering the potential to target solid-state properties that may be influenced by these interactions.
:1, 1:2 or 1:3). These complexes are all ionic in nature, following proton transfer between the acid co-former and DMAN; the extracted proton is held by DMAN in all instances in an intramolecular [N–H⋯N]+ hydrogen bond. A number of structural features are common to all complexes and are found to be tunable in a predictable way using systematic acid co-former substitution. These features include charge-assisted hydrogen bonds formed between acid co-formers in hydrogen bonding motifs consistent with complex stoichiometry, and weak hydrogen bonds which facilitate the crystal packing of DMAN and acid co-former components into a regular motif. Possible crystal structure tuning by co-former substitution can aid the rational design of such materials, offering the potential to target solid-state properties that may be influenced by these interactions.
Introduction
Hydrogen bonds are important for the crystal packing of molecules in the solid state, able to direct molecular configuration and intermolecular interaction.1 Where crystal packing is known to influence properties and function, it is of interest to investigate ways in which molecular association via these interactions can be influenced. Chemical substitution, by modifying molecular building blocks, is one way in which variations in crystal packing have been achieved2–5 and in several cases has led to changes in the properties of solid state materials.6,7Molecular complexes of the proton sponge DMAN (1,8-bis(dimethylamino)naphthalene) with organic acids are an ideal set within which to explore methods of structure tuning. They are regular in their crystallisation behaviour forming very stable ionic complexes8 with common molecular packing motifs.9,10 Such complexes studied previously include DMAN in combination with benzene-1,2,4,5-tetracarboxylic acid, 4,5-di-chlorophthalic acid,11 maleic acid,12 benzene-1,2,3-tricarboxylic acid,13 chloranilic acid,14 1,2-dichloromaleic acid15 and a range of halo benzoic acids.16,17 In these complexes, DMAN typically extracts a proton from the organic acid inducing the formation of O–H⋯O− charge-assisted hydrogen bonds (CAHBs) between acid molecules.11,15,16,18 The extracted proton is held by the DMAN molecule in an asymmetric19–22 intramolecular [N-H⋯N]+ hydrogen bond (IHB).11 The association of the DMAN:acid components in the crystal packing consistently occurs via weak HB interactions in an assembly where the hydrogen bonded acid (ACID−) unit is oriented towards the DMAN cation methyl groups and IHB (Scheme 1).9
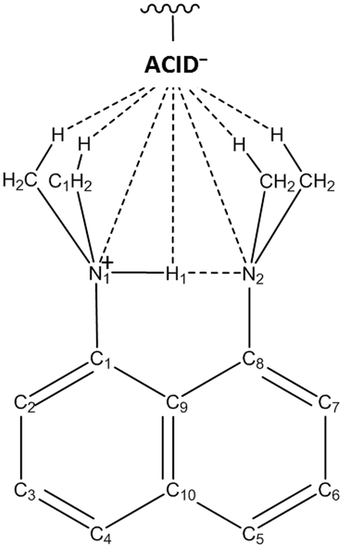 | ||
| Scheme 1 The crystal packing of the DMANH+ cation and ACID− anion via weak hydrogen bonding interactions; the hydrogen bonded acid (ACID−) unit is oriented towards the DMAN cation methyl groups. | ||
For complexes reported to date, modifications to the ACID− component are found to alter both the charge-assisted and weak hydrogen bonding components of the crystal packing.9–11 In this work, acid co-former substitution is demonstrated as a method to tune aspects of these charge-assisted and weak hydrogen bonding interactions in a predictable way including formation of molecular motifs and interaction lengths. Nine molecular complexes of DMAN have been prepared with a range of organic acid co-formers (Scheme 2) substituted with either electron donating (ED) or electron withdrawing (EW) groups.23–26 In this set, there are clear and predictable differences in both the charge-assisted and weak HBs formed on swapping between the ED and EW substituent groups, related to the differing effects of the co-former substituent groups on the acid co-former carboxylate, formed following deprotonation by DMAN.
 | ||
| Scheme 2 1,8-Bis(dimethylamino)naphthalene (DMAN) and organic acid co-formers with substituent groups classed as electron donating ED (circled in blue): 4-hydroxybenzoic acid (1), 3-aminobenzoic acid (2), 4-aminobenzoic acid (3), 3-methoxybenzoic acid (4), and with organic acid co-formers with substituent groups classed as electron-withdrawing EW (circled in orange): 2-cyanobenzoic acid (5), 4-cyanobenzoic acid (6), 4-nitrobenzoic acid (7), 5-nitroisophthalic acid (8) and 2-nitrobenzoic acid (9). | ||
Experimental
Evaporative crystallisation
The molecular complexes were all grown by slow evaporative crystallisation in air, carried out at a range of temperatures.27 Crystallisation stoichiometries of 1:2 DMAN:acid co-former were trialled initially to target the formation of an ACID− dimer as formed commonly in known DMAN benzoic acid molecular complexes.16 Where this 1:2 stoichiometry did not yield single crystals, a 1:1 stoichiometry was trialled. The starting stoichiometry was not always carried through to the molecular complex. Complex 1 (DMAN 4-hydroxybenzoic acid 1:2) was grown from a 1:1 DMAN:acid co-former stoichiometry whilst all other complexes 2–9 were grown from a 1:2 DMAN:acid stoichiometry. 1 was grown from diethyl ether solvent at room temperature. 2 (DMAN 3-aminobenzoic acid 1:2), 3 (DMAN 4-aminobenzoic acid hydrate 3:6:4) and 6 (DMAN 4-cyanobenzoic acid 1:3) were grown from isopropanol solvent at 30 °C, at room temperature and at 50 °C, respectively. 4 (DMAN 3-methoxybenzoic acid 1:2) was grown from acetonitrile solvent at 4 °C. 5 (DMAN 2-cyanobenzoic acid 1:3) was grown from ethyl acetate solvent at 30 °C. 7 (DMAN 4-nitrobenzoic acid 1:3), 8 (DMAN 5-nitroisophthalic acid 1:1) and 9 (DMAN 2-nitrobenzoic acid 1:1), were grown from methanol solvent at 30 °C (8, 9) and 50 °C (7).
Crystallography
For 1, single crystal X-ray diffraction data were collected on beamline I19 in EH1 at Diamond Light Source. For 2, 4–5, 8 and 9 single crystal X-ray diffraction data were collected on beamline 11.3.1 at the Advanced Light Source, U.S.A. For 3 and 6 single crystal X-ray diffraction data were collected at the University of Bath, U.K. For 7, single crystal X-ray diffraction data were collected at the Research Complex at Harwell (RCaH), U.K. The full details of data collection, reduction, crystal structure solution and refinement are given in Table S1.† In nearly all the molecular complexes, hydrogen atoms were located from Fourier difference maps and refined freely. The exception is that of 3 where all amino group hydrogen atoms were refined in calculated positions using HFIX 93 and for C13A, the methyl hydrogen atoms were refined in calculated positions using HFIX 137. Crystallographic data are given in Table 1.:2 (1), DMAN 3-aminobenzoic acid 1:2 (2), DMAN 4-aminobenzoic acid hydrate 3:6:4 (3), DMAN 3-methoxybenzoic acid 1:2 (4), DMAN 2-cyanobenzoic acid 1:3 (5), DMAN 4-cyanobenzoic acid 1:3 (6), DMAN 4-nitrobenzoic acid 1:3 (7), DMAN 5-nitroisophthalic acid 1:1 (8), DMAN 2-nitrobenzoic acid 1:1 (9)
| Complex | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|
| Wavelength (Å)/radiation (Kα) | 0.6889 | 0.8856 | Mo | 0.8856 | 0.8856 | Cu | Cu | 0.8856 | 0.8856 |
| Formula | C28H30N2O6 | C28H32N4O4 | C84H104N12O16 | C30H34N2O6 | C38H33N5O6 | C38H33N5O6 | C35H33N5O12 | C22H23N3O6 | C21H23N3O4 |
| Mol. W (g mol−1) | 490.54 | 488.57 | 1537.79 | 518.59 | 655.69 | 655.69 | 715.66 | 425.43 | 381.42 |
| T (K) | 100 | 100 | 150 | 100 | 150 | 150 | 150 | 100 | 100 |
| Space group | P21/c | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c | P21/c | P21/c |
P |
P21/c | P21/n |
| a (Å) | 11.4729(5) | 9.8631(4) | 13.3902(6) | 9.8917(4) | 11.2829(4) | 21.6378(4) | 14.6269(4) | 7.4488(3) | 10.3320(4) |
| b (Å) | 14.6030(3) | 21.3774(9) | 17.4977(7) | 23.8954(9) | 12.5029(5) | 9.17650(10) | 18.1672(5) | 16.1665(7) | 18.3951(7) |
| c (Å) | 14.7655(5) | 12.5572(5) | 19.3081(9) | 12.0958(5) | 23.4257(10) | 18.3236(3) | 20.9527(6) | 16.8622(8) | 10.3744(4) |
| α (°) | 90 | 90 | 85.639(4) | 90 | 90 | 90 | 93.416(2) | 90 | 90 |
| β (°) | 91.920(3) | 110.968(2) | 70.634(4) | 112.040(2) | 90.718(2) | 112.447(2) | 104.461(3) | 99.229(2) | 103.652(2) |
| γ (°) | 90 | 90 | 69.986(4) | 90 | 90 | 90 | 107.500(3) | 90 | 90 |
| Volume (Å3) | 2472.4(2) | 2472.3(2) | 4006.6(3) | 2650.1(2) | 3304.4(2) | 3362.66(10) | 5086.9(3) | 2004.3(2) | 1916.0(1) |
| Z | 4 | 4 | 2 | 4 | 4 | 4 | 6 | 4 | 4 |
| ρ calc (g cm−3) | 1.318 | 1.313 | 1.275 | 1.300 | 1.318 | 1.295 | 1.402 | 1.410 | 1.322 |
| μ (mm−1) | 0.086 | 0.112 | 0.089 | 0.116 | 0.115 | 0.729 | 0.906 | 0.136 | 0.119 |
| θ range (°) | 1.721–25.502 | 2.374–39.340 | 3.270–27.485 | 2.124–33.661 | 2.167–33.716 | 4.422–71.910 | 3.301–74.703 | 2.188–42.136 | 2.759–41.120 |
| Reflections collected | 21939 |
41246 |
41896 |
37258 |
46488 |
23758 |
37803 |
37365 |
34754 |
| Independent | 5028 | 7542 | 17701 |
5439 | 6806 | 6514 | 20174 |
7291 | 6559 |
| Observed I > 2σ | 3691 | 5867 | 7611 | 3754 | 4353 | 5571 | 16100 |
5697 | 4927 |
| R int | 0.0661 | 0.042 | 0.0645 | 0.0561 | 0.0899 | 0.0306 | 0.0226 | 0.0328 | 0.0525 |
| Completeness (%) | 99.7 | 99.9 | 96.4 | 99.9 | 100.0 | 98.5 | 99.9 | 100.0 | 99.9 |
| Parameters | 445 | 453 | 1271 | 479 | 574 | 574 | 1801 | 372 | 345 |
| GooF | 1.04 | 1.032 | 0.984 | 1.025 | 1.006 | 1.019 | 1.021 | 1.024 | 1.026 |
| R 1 (observed) | 0.0436 | 0.0445 | 0.0793 | 0.045 | 0.0474 | 0.0418 | 0.0396 | 0.0455 | 0.0481 |
| R 1 (all) | 0.0697 | 0.0609 | 0.1953 | 0.0803 | 0.0942 | 0.0487 | 0.0524 | 0.0636 | 0.0713 |
| wR2 (all) | 0.1092 | 0.1215 | 0.2161 | 0.1022 | 0.112 | 0.1117 | 0.1083 | 0.1232 | 0.1266 |
| Δρ (max, min) (e Å−3) | 0.228, −0.229 | 0.437, −0.285 | 0.66, −0.62 | 0.189, −0.275 | 0.277, −0.246 | 0.207, −0.245 | 0.489, −0.423 | 0.463, −0.281 | 0.424, −0.284 |
Results and discussion
In the crystal structures of all molecular complexes (1–9), each symmetry independent DMAN molecule is in its protonated form having extracted a proton from an acid co-former; a DMAN cation (DMANH+) and an acid anion (ACID−) are formed. In all but 9, a neutral acid is present in the crystal structure alongside the ACID− anion; 9 is a 1:1 complex and has a carboxylate group on the single ACID− anion present.
The effect of substituent on acid co-former carboxylate group
The substituent on each acid co-former will have an effect on the stability of the carboxylate group28 formed following deprotonation by DMAN. This is related to the extent to which the substituent groups delocalise the carboxylate negative charge by inductive and/or resonance effects; EW groups increasingly disperse negative charge over the two oxygen atoms whilst ED substituents intensify the negative charge at one site.26,28–30 This effect can be seen in the complexes reported here in the carboxylate C–O− and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond distances of the deprotonated ACID− co-former (Table S2†); these distances are more similar in length for carboxylates with EW substituent groups, indicating a more disperse system, than in those with ED substituent groups, indicating a less disperse system. The extent of delocalisation of the carboxylate negative charge is likely to affect its hydrogen bond propensity.31
O bond distances of the deprotonated ACID− co-former (Table S2†); these distances are more similar in length for carboxylates with EW substituent groups, indicating a more disperse system, than in those with ED substituent groups, indicating a less disperse system. The extent of delocalisation of the carboxylate negative charge is likely to affect its hydrogen bond propensity.31
CAHBs of the ACID− anions
In 1–8, acid co-formers associate via classical O–H⋯O− CAHBs formed between the deprotonated carboxylate group of the ACID− anion and carboxylic acid groups of neighbouring ACID co-formers. This is as found in other DMAN organic acid systems.11,15,16,18 The O–H⋯O− CAHBs (Table S3†) are nearly all asymmetric with respect to the refined hydrogen atom positions. Exceptions to this are the O–H⋯O− CAHBs in 3; the hydrogen atom in these cases is constrained to take a symmetrical position within the HB, lying on a centre of inversion. The refined hydrogen atom positions for all complexes in the CAHBs are supported by the relative bond lengths of the heavy atoms involved in HB formation (Table S2†). The donor–acceptor distances of the O–H⋯O− CAHBs (Table S3†) are in the range of strong hydrogen bonds for all complexes (<2.6 Å);32 for 1–4, they can be classified as short strong CAHBs having donor–acceptor distances less than 2.5 Å.18 The donor–acceptor distances tend to be shorter for the acid co-formers with substituent groups that are ED compared with those that are EW (Fig. S1†). The lower dispersion of the carboxylate negative charge by the ED substituent groups means the carboxylate is a stronger conjugate base.28 In this situation, the electrostatic component of the CAHB will be increased resulting in stronger interactions with shorter donor–acceptor distances. In contrast, in a more disperse system as for the EW substituent groups, the negative charge at each oxygen atom is reduced lowering the electrostatic component of the formed CAHBs;16 the result is weaker interactions with longer O⋯O− donor–acceptor distances.The association of acid co-formers via O–H⋯O− CAHBs in 1–8 generates a range of HB motifs particular to the class of substituent group and complex stoichiometry (Table 2). HB acid dimers (DIMER−) are formed in all the molecular complexes where the acid co-former substituent is ED (1–4); in this motif a deprotonated ACID− and neutral ACID are linked via a single O–H⋯O− CAHB (Fig. 1). 1–4 are also all 1:2 DMAN organic acid molecular complexes. Four symmetry independent acid DIMERs− form in 3. The DIMER− motif occurs where the carboxylate negative charge is more concentrated at a single oxygen atom site by the ED substituent group; this site may then be favoured over the other for CAHB formation. The DIMER− HB motif is seen in similar 1:2 DMAN halobenzoic acid molecular complexes (see Footnote‡).16 There are angular variations between the DIMER− HB motifs (Fig. 1 and Table S4†) with configurations that conform to the classes identified in the crystal structures of a range of DMAN halobenzoic acids.16 The HB DIMERs− in 2–4 adopt the more common pseudo linear conformation, either twisted linear or flat linear, whilst 1 has a less common bent conformation, similarly found in the related materials DMAN 4-bromobenzoic acid and DMAN 4-iodobenzoic acid.16 The DIMER− motifs are isolated and do not catenate further via O–H⋯O− CAHBs. An exception to this is in 1 where the hydroxyl substituent group allows additional O–H⋯O− CAHB links between DIMERs− and a two-dimensional HB sheet results.
| Molecular complex | Substituent group class | HB motif | DMAN:acid co-former stoichiometry |
|---|---|---|---|
| 1 | ED | DIMER− | 1:2 |
| 2 | ED | DIMER− | 1:2 |
| 3 | ED | DIMER− | 1:2 (3:6) |
| 4 | ED | DIMER− | 1:2 |
| 5 | EW | TRIMER− | 1:3 |
| 6 | EW | TRIMER− | 1:3 |
| 7 | EW | TRIMER− | 1:3 (3:9) |
| 8 | EW | (ACID−)n | 1:1 |
| 9 | EW | — | 1:1 |
 | ||
| Fig. 1 The aggregation of ACID− and ACID co-formers into DIMER− HB motifs via a single O–H⋯O− CAHB with configurations bent (1), twisted linear (2 and 4) or flat linear (3: the three symmetry independent DIMERs− are overlaid). | ||
In the molecular complexes where the acid co-former substituent is EW, there are two possibilities for the HB motif. The more common is the formation of HB acid trimers (TRIMER−), where a deprotonated ACID− forms O–H⋯O− CAHBs to two neighbouring acid molecules (Fig. 2). This occurs for all 1:3 DMAN organic acid molecular complexes (5–7); three symmetry independent acid TRIMERs− form in 7. The more disperse negative charge of the carboxylate with an EW substituent means that each oxygen atom may be equally susceptible to CAHB formation33 favouring the TRIMER− motif. In this motif, varying angles between the acid co-former components occur in each molecular complex (Table S5†).
 | ||
| Fig. 2 The aggregation of ACID− and ACID co-formers into TRIMER− hydrogen bonded motifs via two O–H⋯O− CAHBs in complexes 5–7 (the three symmetry independent TRIMERs− are overlaid for 7). | ||
In general, the benzene rings of the co-formers linked by each O–H⋯O− CAHB in the TRIMER− motif are not co-planar, with an angle between the ring planes of approximately 70 to 90°. These TRIMER− motifs are isolated and do not catenate further via O–H⋯O CAHBs. TRIMER− HB motifs are less common for DMAN organic acid molecular complexes and for the association of carboxylate/carboxylic acid groups in general (see Footnote§). An alternative HB motif is formed in 8 where the acid co-former is substituted by a second carboxylic acid group. This is a 1:1 complex. The O–H⋯O− CAHB forms between symmetry related acid co-formers in a one-dimensional HB chain (ACID−)n (Fig. 3). These chains are as found in other 1:1 molecular complexes of DMAN with multi-carboxylic acids including DMAN with benzene-1,2,3-tricarboxylic acid13 and with tartaric acid.18 The benzene rings of the acid co-former molecules in 8, when linked via the O–H⋯O− CAHBs, are significantly non-planar (twisted out of co-planarity by 62°). The donor–acceptor distances of the CAHBs across the molecular complexes here (Table S3 and Fig. S1†) tend to be longer for the TRIMER− HB motifs than when formed in the DIMER− and (ACID−)n HB motifs. This again appears related to the extent of carboxylate negative charge delocalisation; the shortest CAHB distances occur where the carboxylate negative charge is less disperse, indicated by the C–O− and CO bond distances. 9 is a 1:1 complex similar to 8; however, as the co-former lacks a second carboxylic acid group, no O–H⋯O− CAHBs form in the structure following deprotonation by DMAN. It is unknown why 9 forms as a 1:1 complex but it is likely that this stoichiometry prevents behaviour equivalent to the other EW benzoic acid co-formers being observed; as found for the other EW complexes, the carboxylate bond distances in 9 indicate an increasingly delocalised negative charge (dCO 1.248(2) and dC–O 1.254(1) Å) and so a TRIMER− might prevail in a 1:3 version of this complex if this could be synthesised. Both 8 and 9 have EW substituent groups yet form either an alternative (ACID−)n HB motif to the TRIMER− with a shorter CAHB O⋯O− donor–acceptor distance, as in 8, or no CAHB at all, as in 9. In the case of 8, the co-former is a dicarboxylic acid whose electronic structure will be additionally affected by the second carboxylic acid group explaining the differing behaviour. It may be that the predictions made here apply to benzoic acids only.
 | ||
| Fig. 3 The aggregation of symmetry equivalent ACID− co-formers via O–H⋯O− CAHBs into one-dimensional (ACID−)n HB chains in 8. | ||
Weak interactions of the [Me2N–H⋯NMe2]+⋯X− motif
The crystal packing of the ACID− anion HB unit and DMANH+ cation occurs consistently across the molecular complexes reported here via weak HB interactions in an assembly where the ACID− HB unit is oriented towards the DMANH+ cation [N–H⋯N]+ IHB (Scheme 1). Key in this assembly are weak C–H⋯X− HB interactions formed between DMANH+ cation methyl groups and ACID− anion electronegative atoms (X−)9,16,17 generating a [Me2N–H⋯NMe2]+⋯X− motif. This is as found in other ionic DMAN organic acid molecular complexes11–16,18,34,35 and is ubiquitous across the different acid co-former substituent groups. Here, it is notable that in this motif, X− is most often the most electronegative group on the acid co-former which changes depending on substituent group characteristics; it is the carboxylate group for the ED acid co-formers (Fig. 4) but is the substituent group for EW acid co-formers (Fig. 5). Being a stronger conjugate base,28 the ED acid co-former carboxylate groups will be more strongly attracted to the [Me2N–H⋯NMe2]+ fragment, favouring the assembly shown in Fig. 4. In contrast, the weaker conjugate basicity of the EW acid co-former carboxylate group means it may be more favourable for the more electronegative substituent group to orient towards the [Me2N–H⋯NMe2]+ fragment, explaining why this has been found for the EW groups in this study (see Footnote¶).Weak C–H⋯O− HBs form the [Me2N–H⋯NMe2]+⋯X− motif where X− is the carboxylate group; there is also variation in the orientation of the acid anion DIMER− HB unit towards the [Me2N–H⋯NMe2]+ fragment (Fig. 4). The DIMER− has either a bisecting approach to the IHB, as in 2 and 4, or a flat approach, as in 1 and 3. The weak C–H⋯X− HB interactions in the [Me2N–H⋯NMe2]+⋯X− motif vary with the different nature of the available HB acceptors on the X− substituent group; these are C–H⋯N HBs in 5 and 6 and C–H⋯O HBs in 8 and 7. In this motif, the EW substituent group approaches the [Me2N–H⋯NMe2]+ fragment either side-on or tail-on (Fig. 5).
 | ||
| Fig. 4 The approach of the DMANH+ cation (blue motif) by the ACID− anion units (purple/orange motif): the ACID− anion carboxylate group is oriented towards the [Me2N–H⋯NMe2]+ fragment in 1–4 and 9 (the three symmetry independent assemblies are shown for 3). | ||
 | ||
| Fig. 5 The approach of the DMANH+ cation (blue motif) by the ACID− anion HB units (purple/orange motif): the EW substituent group is oriented towards the [Me2N–H⋯NMe2]+ fragment either side-on (5 and 8) or tail-on (6 and 7: the three symmetry independent assemblies are shown for 7). | ||
9 exhibits anomalous behaviour here. Despite the substituent group being EW, the less electronegative carboxylate group is oriented towards the [Me2N–H⋯NMe2]+ fragment and the weak interactions are C–H⋯O− HBs. This may be due to the 1:1 stoichiometry of components for this system; the carboxylate group is oriented towards the [Me2N–H⋯NMe2]+ fragment where there is no additional co-former molecule to stabilise its negative charge, unlike in the other molecular complexes here.
The lengths of the weak C–H⋯X− interactions across the complexes are generally shorter where X− is the carboxylate than where X− is the EW substituent group (Tables S6 and S7, Fig. S2†). Unlike the carboxylate group, the EW substituent is not formally charged; in this situation the electrostatic component of the weak C–H⋯X− HB interactions is reduced resulting in longer C⋯X− distances (the positively charged [Me2N–H⋯NMe2]+ fragment interacts with a weaker partial charge on the EW substituent group).36 The weak C–H⋯X− HB interactions also tend to be shortest to the protonated dimethylamino (Me2N1) (Fig. S2†); this is in line with the previously proposed multicentre character of [Me2N–H⋯NMe2]+⋯X− where weak minor interactions with X− can cause localisation of the proton in the [N–H⋯N]+ intramolecular IHB.9
Steric influence on weak interactions in [Me2N–H⋯NMe2]+⋯X− motifs
The sterics of the carboxylate group may also be a determining factor in the formation of the weak interactions in this set. The carboxylate group tends to form a single CAHB in a DIMER− motif for ED substituent groups and two CAHBs in a TRIMER− HB motif for EW substituent groups. The bulky TRIMER− and the additional CAHB of the carboxylate group when in this HB motif may sterically hinder further interaction of this group with the [Me2N–H⋯NMe2]+ fragment, impeding its approach; the substituent group is oriented instead towards this fragment. Sterics may then also explain why in 9 the less electronegative carboxylate group is oriented towards the [Me2N–H⋯NMe2]+ fragment, as shown in Fig. 4; the carboxylate group is unimpeded by CAHBs and is a smaller molecular fragment for the approach.The DMANH+ cation
In the crystal structures of each molecular complex, DMAN molecules are present in their protonated form (DMANH+) only. This is widely found in DMAN organic acid molecular complexes; the crystallisation of neutral DMAN molecules alongside DMANH+ cations is rare, found in very few instances.16 In all the molecular complexes reported here, the proton extracted from the acid co-formers is held by the DMANH+ cation in a short strong [N–H⋯N]+ intramolecular hydrogen bond (IHB). The IHB N⋯N distances (Table S8†) are in the range of those found previously.37 They tend to be more consistent in length in the DMANH+ cations where the acid co-former is ED (dN⋯N 2.56 to 2.58 Å) than where it is EW (dN⋯N 2.55 to 2.62 Å). The N⋯N distance in 9 is an outlier for this set, being significantly longer than in 1–8. N⋯N IHB distances longer than 2.60 Å are less common though not unknown in these types of complexes.13,15,18The refined hydrogen atom positions in the IHBs indicate an asymmetrically located proton in 2, 3 (DMAN H1C), 4, 6, 7 and 9 whilst symmetric positions are indicated in 1, 3 (DMANH1A and DMANH1B) and 8. Here, the refined positions are supported by the C–N distances and structural parameters of the DMANH+ molecules (Tables S9 and S10†). An asymmetry in the position of the IHB proton is mirrored by an asymmetry in the C–N distances and structural parameters on either side of the DMANH+ cation.
Weak interactions dominate the packing of the DMANH+ cations in the molecular complexes reported here. Contacts between the cations include weak π–π and/or methyl⋯π interactions involving DMANH+ methyl and naphthalene ring groups. DMANH+ packing motifs vary on co-former substitution (Fig. 6). Packing of cations occurs anti-parallel in pairs with varying degrees of overlap, either as an isolated stack (2, 4), an offset pair (7) or in a continuous column (3, 8, 9). Alternatively, in several cases (1, 5, 6) DMANH+ cations pack in one-dimensional chains through the structure. Stoichiometry, rather than substituent group, has an effect on DMANH+ packing, as found elsewhere;16 here it can be seen that the cations in the 1:1 and 1:2 complexes have a tendency to stacking which is not seen in the 1:3 complexes in which cation packing is more flat through the structure.
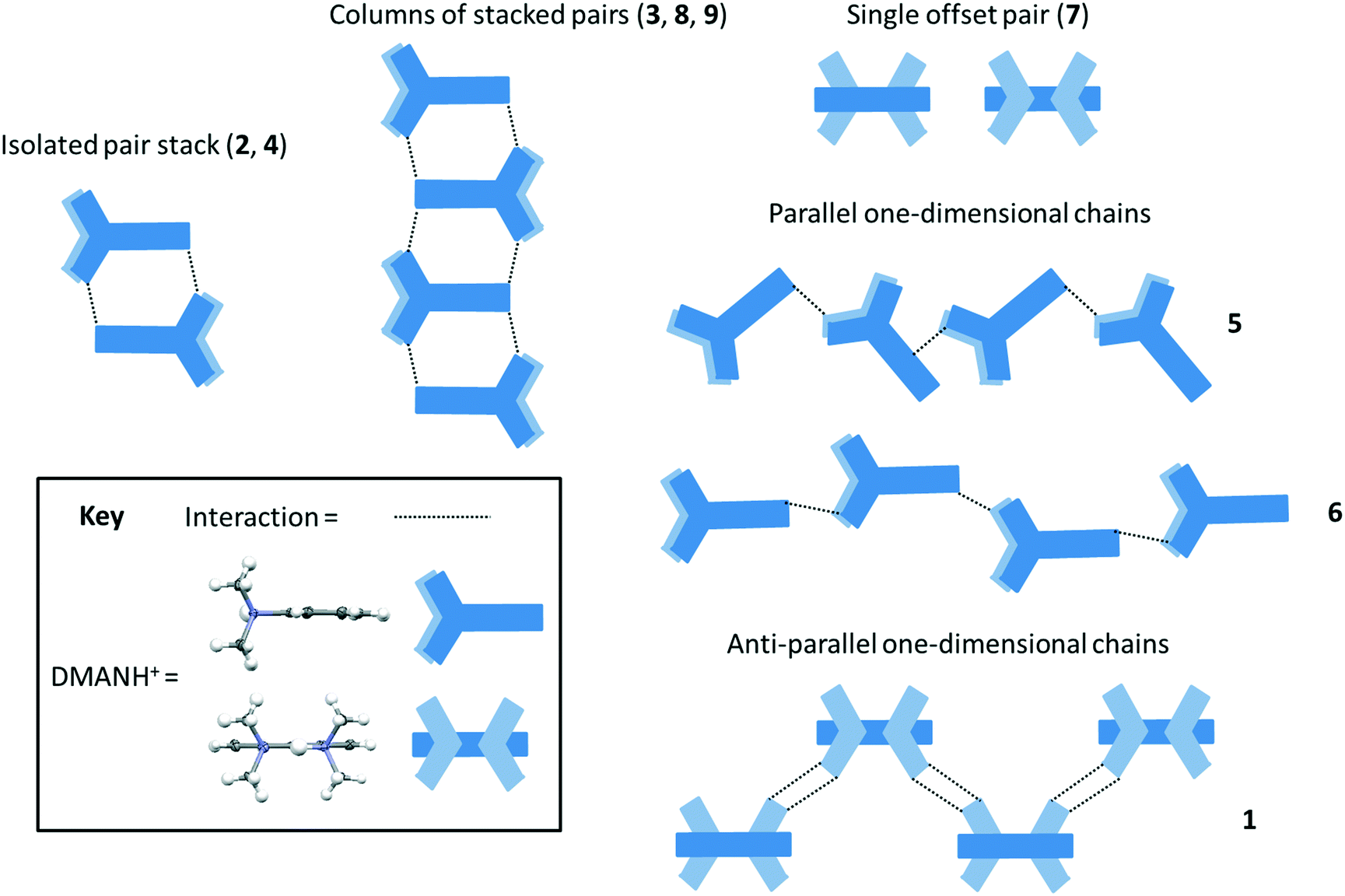 | ||
| Fig. 6 Molecular packing of the DMANH+ cations (blue motif): anti-parallel in pairs in an isolated stack (2, 4) or in columns (3, 8 and 9), in offset pairs (7), in one-dimensional chains either parallel with varying degrees of co-planarity (5 and 6) or anti-parallel (1). | ||
Conclusions
In this work, nine molecular complexes of DMAN have been prepared with a range of substituted benzoic acid co-formers. A systematic study of selected charge-assisted and weak hydrogen bonding interactions common to all molecular complexes has revealed how co-former substitution may be used to tune such interactions. The electronic properties of the acid co-former substituent group, whether electron donating or withdrawing, are found to be important in influencing the formation of these interactions, in terms of molecular motifs, and their interaction distances. Finding ways to influence the formation of intermolecular interactions represents progress in materials design allowing structural features to be predictably accessed. Importantly, where structure affects property, the tuning of intermolecular interactions can have implications in host–guest chemistry31 and accessing proton transfer behaviour;17,38,39 the latter offers the potential for colour change40,41 or ferroelectric42,43 properties in a material.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge the support from the University of Bath and Diamond Light Source Ltd. for a studentship to L. K. S. C. C. W. and P. R. R. are grateful to the EPSRC for continued support (EP/K004956/1). We thank Diamond Light Source for access to beamline I19 (MT-8803) for data for sample 1. We would also like to thank the Advanced Light Source, LBNL for beamtime to collect on samples 2, 4–5, 8, 9 and S. J. Teat and K. J. Gagnon for their assistance. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under contract no. DE-AC02-05CH11231.Notes and references
- M. C. Etter, Z. Urbanczyk-Lipkowska, M. Zia-Ebrahimi and T. W. Panunto, J. Am. Chem. Soc., 1990, 112, 8415–8426 CrossRef.
- D. Vuk, K. Molčanov and I. Škorić, J. Mol. Struct., 2014, 1068, 124–129 CrossRef.
- L. J. McCormick, C. McDonnell-Worth, J. A. Platts, A. J. Edwards and D. R. Turner, Chem. – Asian J., 2013, 8, 2642–2651 CrossRef PubMed.
- A. Cvetkovski, V. Bertolasi and V. Ferretti, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 326–334 Search PubMed.
- J.-H. Dou, Y.-Q. Zheng, Z.-F. Yao, Z.-A. Yu, T. Lei, X. Shen, X.-Y. Luo, J. Sun, S.-D. Zhang, Y.-F. Ding, G. Han, Y. Yi, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2015, 137, 15947–15956 CrossRef PubMed.
- Y.-X. Li, X.-F. Yang, J.-L. Miao, Z.-W. Zhang and G.-X. Sun, CrystEngComm, 2016, 18, 2098–2104 RSC.
- B. Kupcewicz and M. Małecka, Cryst. Growth Des., 2015, 15, 3893–3904 Search PubMed.
- A. L. Llamas-Saiz, C. Foces-Foces and J. Elguero, J. Mol. Struct., 1994, 328, 297–323 CrossRef.
- K. Wozniak, P. R. Mallinson, G. T. Smith, C. C. Wilson and E. Grech, J. Phys. Org. Chem., 2003, 16, 764–771 CrossRef.
- G. S. Nichol and W. Clegg, Cryst. Growth Des., 2006, 6, 451–460 Search PubMed.
- P. R. Mallinson, G. T. Smith, C. C. Wilson, E. Grech and K. Wozniak, J. Am. Chem. Soc., 2003, 125, 4259–4270 CrossRef PubMed.
- E. Bartoszak, Z. Dega-Szafran, M. Grundwald-Wyspianska, M. Jaskolski and M. Szafran, J. Chem. Soc., Faraday Trans., 1993, 89, 2085–2094 RSC.
- M. L. Raves, J. A. Kanters and E. Grech, J. Mol. Struct., 1992, 271, 109–118 CrossRef.
- J. A. Kanters, A. Schouten, A. J. M. Duisenberg, T. Glowiak, Z. Malarski, L. Sobczyk and E. Grech, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, 47, 2148–2151 CrossRef.
- K. Wozniak, C. C. Wilson, K. S. Knight, W. Jones and E. Grech, Acta Crystallogr., Sect. B: Struct. Sci., 1996, 52, 691–696 CrossRef.
- L. H. Thomas, A. O. F. Jones, A. A. Kallay, G. J. McIntyre and C. C. Wilson, Cryst. Growth Des., 2016, 16, 2112–2122 Search PubMed.
- A. O. F. Jones, A. A. Kallay, H. Lloyd, G. J. McIntyre, C. C. Wilson and L. H. Thomas, Cryst. Growth Des., 2016, 16, 2123–2129 Search PubMed.
- O. R. Israël, J. A. Kanters and E. Grech, J. Mol. Struct., 1992, 274, 151–162 CrossRef.
- J. A. Platts, S. T. Howard and K. Wozniak, J. Org. Chem., 1994, 59, 4647–4651 CrossRef.
- K. Woźniak, T. M. Krygowski, B. Kariuki, W. Jones and E. Grech, J. Mol. Struct., 1990, 240, 111–118 CrossRef.
- C. L. Perrin and B. K. Ohta, J. Am. Chem. Soc., 2001, 123, 6520–6526 CrossRef PubMed.
- L. Sobczyk, J. Mol. Struct., 2010, 972, 59–63 CrossRef.
- A. Jacobs, Understanding Organic Chemistry Reactions, Cambridge University Press, Cambridge, 1997 Search PubMed.
- P. W. G. Smith and A. R. Tatchell, Aromatic Chemistry: Organic Chemistry for General Degree Students, First edn., Pergamon Press, Oxford, 1969 Search PubMed.
- P. M. Dewick, Essentials of Organic Chemistry: For Students of Pharmacy, Medicinal Chemistry and Biological Chemistry, John Wiley & Sons, Ltd, Chichester, 2006 Search PubMed.
- M. A. Hernandez and A. Rathinavelu, Basic Pharmacology Understanding Drug Actions and Reactions, CRC Press, Boca Raton, 2006 Search PubMed.
- L. K. Saunders, Synchrotron X-ray Diffraction Studies of Proton Transfer in Hydrogen Bonded Molecular Complexes, University of Bath, U.K., 2016 Search PubMed.
- T. L. Lemke, Review of Organic Functional Groups: Introduction to Medicinal Organic Chemistry, Lippincott Williams & Wilkins, Baltimore, 2003 Search PubMed.
- C. P. Murthy, P. K. Dubey, D. Ashok and S. F. Mehdi Ali, University Chemistry Volume - II, University Chemistry, New Age International (P) Ltd., Publishers, New Delhi, 1996 Search PubMed.
- S. Agarwal, Engineering Chemistry Fundamentals and Applications, Cambridge University Press, Delhi, 2015 Search PubMed.
- I. L. Kirby, M. Brightwell, M. B. Pitak, C. Wilson, S. J. Coles and P. A. Gale, Phys. Chem. Chem. Phys., 2014, 16, 10943–10958 RSC.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, New York, 1997 Search PubMed.
- G. L. Patrick, An Introduction to Drug Synthesis, Oxford University Press, Oxford, 2015 Search PubMed.
- R. T. Stibrany and J. A. Potenza, CSD Communication, 2006 Search PubMed.
- P. R. Mallinson, K. Woźniak, C. C. Wilson, K. L. McCormack and D. S. Yufit, J. Am. Chem. Soc., 1999, 121, 4640–4646 CrossRef.
- M. Miljkovic, Electrostatic and Stereoelectronic Effects in Carbohydrate Chemistry, Springer, New York, 2014 Search PubMed.
- I. Majerz and I. Olovsson, Phys. Chem. Chem. Phys., 2009, 11, 1297–1302 RSC.
- A. O. F. Jones, N. Blagden, G. J. McIntyre, A. Parkin, C. C. Seaton, L. H. Thomas and C. C. Wilson, Cryst. Growth Des., 2013, 13, 497–509 Search PubMed.
- A. O. F. Jones, M.-H. Lemee-Cailleau, D. M. S. Martins, G. J. McIntyre, I. D. H. Oswald, C. R. Pulham, C. K. Spanswick, L. H. Thomas and C. C. Wilson, Phys. Chem. Chem. Phys., 2012, 14, 13273–13283 RSC.
- M. T. Reetz, S. Höger and K. Harms, Angew. Chem., Int. Ed. Engl., 1994, 33, 181–183 CrossRef.
- C. L. Jones, C. C. Wilson and L. H. Thomas, CrystEngComm, 2014, 16, 5849–5858 RSC.
- K. Lee, B. Kolb, T. Thonhauser, D. Vanderbilt and D. C. Langreth, Phys. Rev. B, 2012, 86, 104102 CrossRef.
- S. Horiuchi and Y. Tokura, Nat. Mater., 2008, 7, 357 CrossRef PubMed.
- F. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystal structure refinement details; selected molecular geometry tables, selected hydrogen bond tables, fingerprint plots/Hirshfeld surfaces. CCDC 1831638–1831646. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ce00443a |
| ‡ In related molecular complexes of DMAN with halobenzoic acids,16 where the substituent group is a halogen, the acid DIMER− HB motifs prevail. Halogen substituents are electron withdrawing by induction but have similar resonance effects to ED substituent groups.26 This may be why DIMER− HB motifs are found in these complexes; the carboxylate C–O and CO bond lengths are also very different in length16 as found in the acid co-formers here with ED substituents. |
| § A survey of the Cambridge Structural Database (CSD, version 5.38 November 2016)44 for carboxyl carboxylate catemers in organic only systems (based on O–H⋯O− contacts between RCO2− and either a single RCO2H forming a DIMER− or to two RCO2H forming a TRIMER−, where R is any group) indicated that catenated DIMERs− were more prevalent occurring in 1653 instances in contrast to 302 instances for TRIMERs−. |
| ¶ In previously reported molecular complexes of DMAN with halobenzoic acids,16 X− is also the carboxylate group despite the halogen substituent groups being more electronegative. |
| This journal is © The Royal Society of Chemistry 2018 |