
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConformational chiral polymorphism in cis-bis-triphenylphosphine complexes of transition metals†
Never
Tshabang
*a,
Gaone P.
Makgatle
a,
Susan A.
Bourne
 *b,
Nina
Kann
c,
Jack D.
Evans‡
d,
François-Xavier
Coudert
*d and
Lars
Öhrström
*c
*b,
Nina
Kann
c,
Jack D.
Evans‡
d,
François-Xavier
Coudert
*d and
Lars
Öhrström
*c
aDepartment of Chemistry, Faculty of Science, University of Botswana, Gaborone, Botswana. E-mail: tshabang@mopipi.ub.bw
bCentre for Supramolecular Chemistry Research, Department of Chemistry, University of Cape Town, Rondebosch 7701, Cape Town, South Africa. E-mail: susan.bourne@uct.ac.za
cChemistry and Biochemistry, Dept. of Chemistry and Chemical Engineering, Chalmers University of Technology, SE-41296 Göteborg, Sweden. E-mail: ohrstrom@chalmers.se
dChimie ParisTech, PSL University, CNRS, Institut de Recherche de Chimie Paris, 75005 Paris, France. E-mail: fx.coudert@chimieparistech.psl.eu
First published on 8th June 2018
Abstract
The structure of cis-[Mo(CO)4(PPh3)2] 1 was determined by F. A. Cotton, D. J. Darensbourg, S. Klein and B. W. S. Kolthammer, Inorg. Chem., 1982, 21, 1651–1655, with the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . A second polymorph 2 is reported here, with the space group P21/c. The compounds differ in the interactions between the conformational chiral triphenylphosphine groups. In 1, there is π–π stacking between adjacent phenyl groups, whereas in 2, there are σ–π interactions instead. A search of the Cambridge Structural Database reveals that this is a relatively frequent occurrence in cis-bis-triphenylphosphine complexes and the phenomenon can be analysed by means of the C(ipso)–P–M–P torsion angles. The majority of compounds fall in the π–π stacking data area with torsion angles of 10–15° and 55–60°; however, for octahedrally coordinated metals, the optimum is a σ–π interaction at 40°/40°. This corresponds well to the values in 2: 46°/40°, but for 1, we instead find the torsion angles to be 11°/18°. There is indeed a small occurrence of these values as well in the data, and it appears that for 1, this conformation is stabilised by weak CO⋯H–C hydrogen bonds. Density functional theory (DFT) calculations indicate that 1 is the more stable polymorph by 72 kJ mol−1 but that the strain of the complexes (the difference between a relaxed molecule in the respective conformation and the structure in the crystal) is larger for 1 than for 2, further indicating that a special intermolecular interaction is responsible for the stability of this polymorph. In both polymorphs, the triphenylphosphines have the same conformational chirality, consistent with single-molecule calculations that predict racemic conformations to be substantially higher in energy for both σ–π interactions (+17 kJ mol−1) and π–π stacking (+30 kJ mol−1).
. A second polymorph 2 is reported here, with the space group P21/c. The compounds differ in the interactions between the conformational chiral triphenylphosphine groups. In 1, there is π–π stacking between adjacent phenyl groups, whereas in 2, there are σ–π interactions instead. A search of the Cambridge Structural Database reveals that this is a relatively frequent occurrence in cis-bis-triphenylphosphine complexes and the phenomenon can be analysed by means of the C(ipso)–P–M–P torsion angles. The majority of compounds fall in the π–π stacking data area with torsion angles of 10–15° and 55–60°; however, for octahedrally coordinated metals, the optimum is a σ–π interaction at 40°/40°. This corresponds well to the values in 2: 46°/40°, but for 1, we instead find the torsion angles to be 11°/18°. There is indeed a small occurrence of these values as well in the data, and it appears that for 1, this conformation is stabilised by weak CO⋯H–C hydrogen bonds. Density functional theory (DFT) calculations indicate that 1 is the more stable polymorph by 72 kJ mol−1 but that the strain of the complexes (the difference between a relaxed molecule in the respective conformation and the structure in the crystal) is larger for 1 than for 2, further indicating that a special intermolecular interaction is responsible for the stability of this polymorph. In both polymorphs, the triphenylphosphines have the same conformational chirality, consistent with single-molecule calculations that predict racemic conformations to be substantially higher in energy for both σ–π interactions (+17 kJ mol−1) and π–π stacking (+30 kJ mol−1).
1. Introduction
Triarylphosphines are ubiquitous in organometallic chemistry. As early as 1948, Reppe used [NiBr2(PPh3)2],2 and ever since the preparation of Wilkinson's catalyst, [RhCl(PPh3)3], in 1965 (ref. 3) and the subsequent employment of triphenylphosphine and its derivatives as ligands to transition metal catalysts in industrial processes, for example hydroformylation,4 they have been trustworthy work horses for the organometallic and catalysis community.5PPh3 continues to play an important role in catalysis to this day; examples include the use of [Pd(PPh3)4] in classical cross-coupling reactions with aryl halides,6 applications of diphosphine complexes such as [PdCl2(PPh3)2] in carbonylative Sonogashira reactions,7,8 and as a [RuCl(phenpy-OH)(PPh3)2]PF6 catalyst in the β-alkylation of secondary alcohols with primary alcohols.9 Looking at other metal-PPh3 complexes, iron catalysts such as [Fe(H)(CO)(NO)(PPh3)2] and [Fe(CO)Cp(PPh3)I] have been applied in the hydrosilylation of alkynes10 and acetophenone,11 respectively, while cobalt complexes like [Co(H)(N2)(PPh3)3] or [CoH3(PPh3)3] can effect CH-activation of aromatic compounds.12 A recent application of interest involves the dual use of [Ph3PAuCl] and visible light to achieve the synthesis of biarylic compounds via a Suzuki-type coupling.13 Triphenylphosphine has also been employed in itself as an organocatalyst in reactions of allenes with electrophiles,14 as well as in [2 + 2 + 2]-annulations to form dihydropyridine structures.15
That triarylphosphines are conformationally chiral was realised long ago,16,17 and they have been subjected to detailed studies,18 but such analyses of bis-triphenylphosphine complexes are scarce. In this communication, we report on a new polymorph of cis-[Mo(CO)4(PPh3)2] 2 in the space group P21/c, distinct from the earlier reported P polymorph 1 in terms of both phosphine configurations and intermolecular interactions. We have also placed this finding in a broader context by analysing diphosphine complexes in the Cambridge Structural Database (CSD) and performing quantum chemical calculations on both crystals and single molecules.
In principle, one could envisage four different ways in which these molecules could vary in their conformations. We could think of either σ–π interactions or π–π stacking as potentially being the most favoured intramolecular interaction between the closest phenyl groups of the two triphenylphosphine units. Added to this is the possibility of having either homo (ΔΔ or ΛΛ) or heterochiral triphenylphosphines, giving a total of four possible conformers: σπ–ΔΔ, σπ–ΔΛ, ππ–ΔΔ, and ππ–ΔΛ. Considering also the possibility of different packings, the potential for polymorphism seems rather large. However, it is quite possible that some of the conformers have too high energies to be accessible, even if a favoured packing can be arranged.
2. Experimental
2.1 Materials and methods
The structure determination details are found in Table 1, and an ORTEP type drawing for the molecular unit is shown in Fig. 1.
| 1 | 2 | |
|---|---|---|
| a Cotton et al.1 | ||
| Empirical formula | C40H30MoO4P2 | C40H30MoO4P2 |
| Molecular mass (g mol−1) | 732.52 | 732.52 |
| Crystal size (mm) | 0.03 × 0.09 × 0.16 | |
| Temperature of data collection | 298 | 173(2) |
| Crystal symmetry | Triclinic | Monoclinic |
| Space group |
P |
P21/c |
| a (Å) | 11.522(1) | 9.4289(7) |
| b (Å) | 16.909(3) | 38.395(3) |
| c (Å) | 9.633(2) | 9.9447(7) |
| α (°) | 98.05(2) | 90 |
| β (°) | 110.29(1) | 107.876(1) |
| γ (°) | 99.95(1) | 90 |
| Z | 2 | 4 |
| Volume (Å3) | 1692.73(4) | 3426.4(4) |
| Densitycalc (g cm−3) | 1.437 | 1.420 |
| 2θ range scanned (°) | 2.12–27.95 | |
| F (000) | 1496 | |
| No. of reflections collected | 57![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 328 328 |
|
| No. of unique reflections | 8215 | |
| No. of reflections with I > 2θ | 6229 | |
| Goodness of fit, S | 1.016 | |
| R 1 (I > 2σI) | 0.043 | 0.0392 |
| Final wR2 (all data) | 0.0857 | |
| Min, max e density/e | −0.470, 0.540 | |
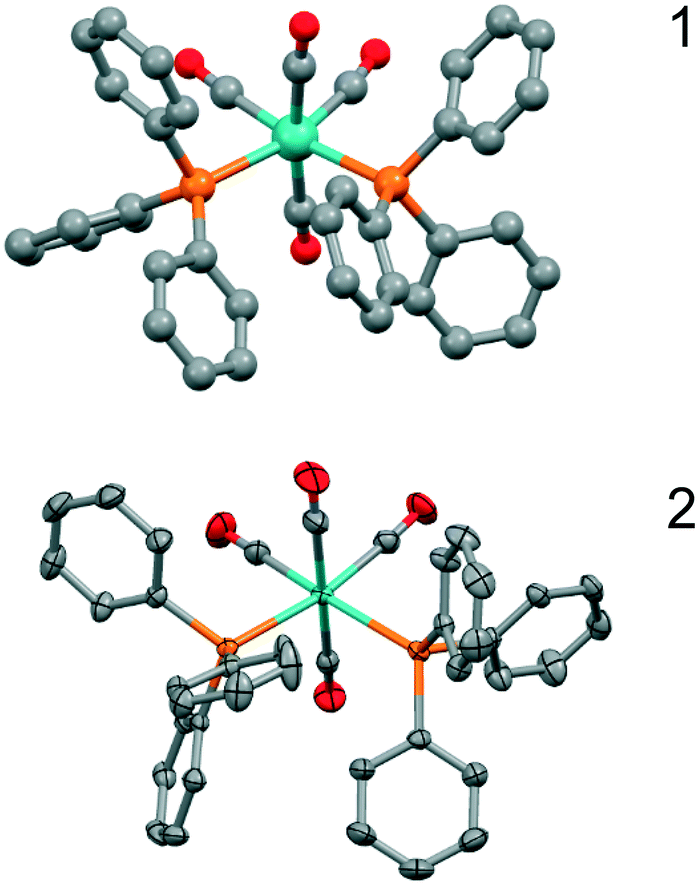 | ||
| Fig. 1 Drawings of the molecular units of 1 (top, ball-and-stick) and 2 (bottom, ORTEP type). Hydrogen atoms are omitted for clarity. | ||
2.2 Synthesis
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O) 2013, 1899, and 1877. 1H NMR (CD2Cl2, 300.13 MHz) δ: 7.26–7.41 (m, 30 H, Ph) ppm. 31P NMR (CD2Cl2, 75.47 MHz); δ = 37.7 ppm. Single crystals of the compound, suitable for X-ray crystallographic analysis, were obtained by slow diffusion of hexane into a solution of 2 in dichloromethane for a period of one week. The crystals were sent to the University of Cape Town for X-ray crystallographic analysis.
O) 2013, 1899, and 1877. 1H NMR (CD2Cl2, 300.13 MHz) δ: 7.26–7.41 (m, 30 H, Ph) ppm. 31P NMR (CD2Cl2, 75.47 MHz); δ = 37.7 ppm. Single crystals of the compound, suitable for X-ray crystallographic analysis, were obtained by slow diffusion of hexane into a solution of 2 in dichloromethane for a period of one week. The crystals were sent to the University of Cape Town for X-ray crystallographic analysis.
3. Results and discussion
3.1 Synthesis
Crystals of cis-[Mo(CO)4(PPh3)2] in the space group P21/c2 were obtained when the synthesis of fac-[Mo(CO)3(PPh3)3] was attempted by the method for preparing fac-[Cr(CO)3(PPh3)3] reported by Nicholls and Whiting.24 Crystals suitable for single crystal X-ray diffraction were obtained by slow diffusion of hexane into a solution of 2 in dichloromethane for a period of one week. It should be noted that the previously reported crystals 1 of cis-[Mo(CO)4(PPh3)2] in the space group P were obtained by recrystallizations from a chloroform/methanol mixture at 0 °C.1 This underpins the important role of the solvent during crystallisation.
3.2 X-ray crystallography structure analysis
The structure determination details are displayed in Table 1 and an ORTEP type drawing for the molecular unit is shown in Fig. 1.On the bonding level, 1 and 2 are very similar as the Mo–C and Mo–P bonds are close (Mo–P, 2.58 Å vs. 2.58 Å, Mo–C, 1.98–2.04 Å vs. 1.97–2.06 Å), but the configurations of the closest phenyl contacts between triphenylphosphine ligands are distinctively different, as shown schematically in Fig. 2, corresponding to the π–π- and σ–π-cases.
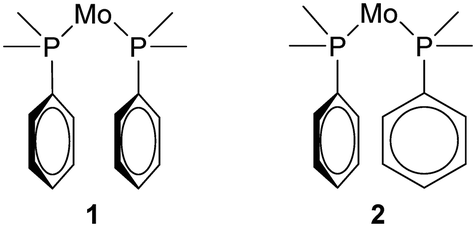 | ||
| Fig. 2 Schematic difference between the bis-phosphine units in 1 and 2. | ||
However, this is not a question of the two polymorphs having, in one case, the same configurational chirality on the phosphine ligands and the other one having opposite chiralities. In both cases, as far as can be quantified, the chirality is the same for the two phosphine ligands, but in neither case, the propeller-like chirality is perfect. Thus, what we have are the σπ–ΔΔ and ππ–ΔΔ conformers. In fact, judging from the physical ball-and-spoke molecular models, it seems difficult to have different chiralities on the two PPh3 ligands and turn these into a sensible conformation that does not generate very close contacts. This, however, needs to be quantified and confirmed by quantum chemical calculations.
3.3 Structure calculations
To gain some insight into the factors playing a role in this polymorphism case, DFT calculations were performed, both under periodic boundary conditions on the two crystal systems, single molecule calculations for the chiral conformers 1 and 2, and the corresponding hypothetical non-chiral conformers 3 and 4.The results in terms of energy are summarised in Table 2. First, we note that the small difference on a single molecule level for 1 and 2 is just about significant, as the two configurations will differ only because of relatively weak interactions between the phenyl rings. Second, on a structural level, the two polymorphs are well reproduced by the calculations (see Fig. 3).
| Space group | Conformation | Relative crystal energy (kJ mol−1) | Lattice energya (kJ mol−1) | Packing energyb (kJ mol−1) | Molecular strain in crystalc (kJ mol−1) | Single molecule energy difference (kJ mol−1) | |
|---|---|---|---|---|---|---|---|
| a Lattice energy is the difference in energy between the crystal and the free molecule in its relaxed form. b Packing energy is the difference in energy between the crystal and the individual molecule in the same conformation as in the crystal. c Lattice energy = packing energy + strain energy from the deformation of the molecule to fit into the crystal lattice. | |||||||
| 1 |
P |
ππ–ΔΔ | 0 | −245.6 | −282.9 | 37.3 | 0 |
| 2 | P21/c | σπ–ΔΔ | 72.4 | −235.6 | −260.6 | 25.0 | 7.7 |
| 3 | n.a. | σπ–ΔΛ | 16.7 | ||||
| 4 | n.a. | ππ–ΔΛ | 30.3 | ||||
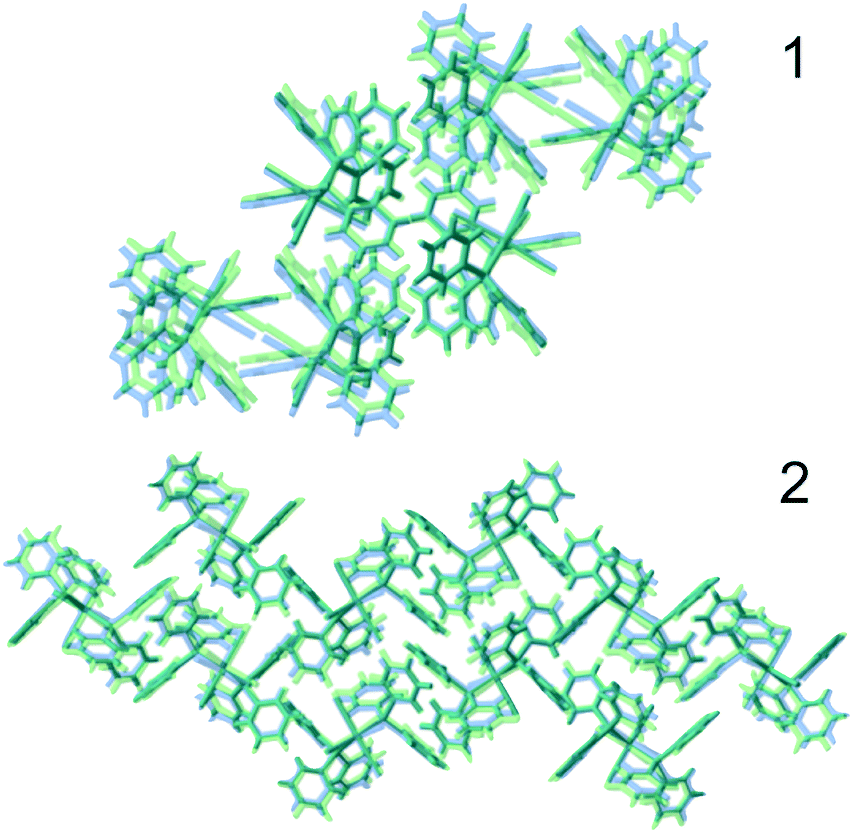 | ||
| Fig. 3 Comparison of experimental (blue) and optimised structures (green) of 1 (top) and 2 (bottom). | ||
The four molecular conformers 1–4 are displayed in Fig. 4.
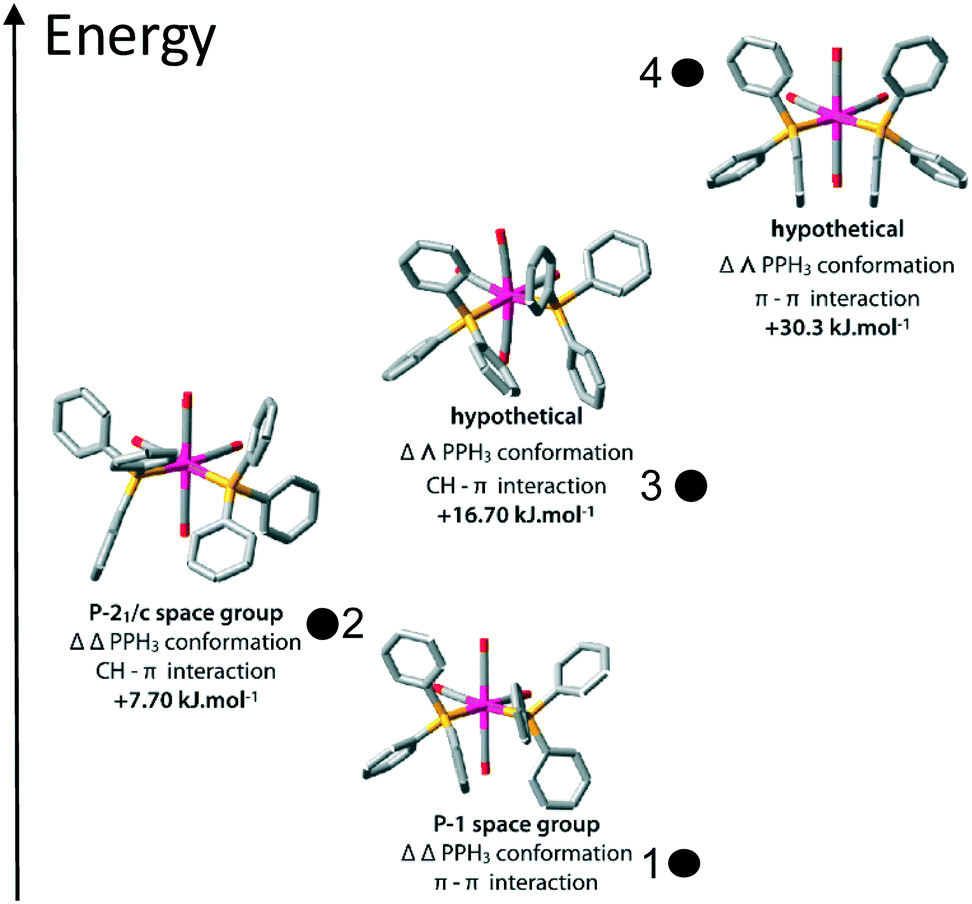 | ||
| Fig. 4 Optimized structures of the four conformations of cis-[Mo(CO)4(PPh3)2]. The two lowest energy conformers correspond to the experimentally observed compounds 1 and 2. | ||
On a crystal level, 1 is the more stable polymorph, consistent with its higher density (Table 1). There may also be more specific interactions that evoke a more stable polymorph. Hirschfeld surface analysis25–27 (Fig. 5) indicates one fairly strong double CO⋯H interaction at 2.135 Å, O⋯C at 3.190 Å, and CHO at 163.8° (calc.) and 2.5404 Å, 3.490 Å, and 177.7° (exp.). These are short, and C⋯O (the best determined distance) is very close to the optimum C–H⋯OC distance which is 3.48 Å according to analysis of data in the CSD. However, the optimum angle is 116°, which is very far from the observed 177.7° (see the ESI,† Fig. S1).
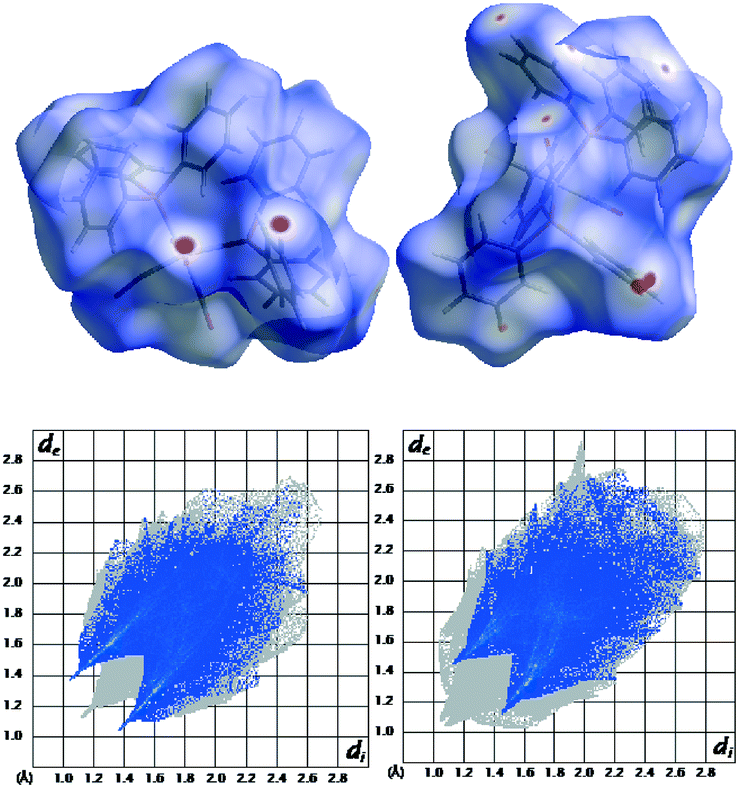 | ||
| Fig. 5 Top: Hirshfeld surface analysis of 1 (left) and 2 (right), bottom: Hirshfeld surface analysis fingerprint plots of 1 (left) and 2 (right); O⋯H interactions are emphasised. The perspectives are different to show the major interactions for both compounds. | ||
The fingerprint plot also reveals a higher H⋯H repulsion in 2 (the peaks pointing to the lower left along the diagonal). However, the strain induced in the two conformers going from the free single molecules to the crystal is larger by 12.3 kJ mol−1 for 1. This can also be quantified on a structural level as the total difference between the Cipso–P–Mo–P torsion angles between the optimised free molecule and the molecule restricted in the crystal is almost double for 1 compared to 2 (see Table 3).
| Comp. | Space gr. | In crystal | Free molecule | Total abs. diff. free crystal | Ph⋯Ph in crystal | Ph⋯Ph free molecule | ||
|---|---|---|---|---|---|---|---|---|
| φ 1 (°) | φ 2 (°) | φ 1 (°) | φ 2 (°) | β (°) | β (°) | |||
| 1 |
P |
11.1 | 17.8 | n.a. | n.a. | 17.0 | n.a. | |
| 1 |
P DFT |
10.0 | 17.1 | 20.1 | 20.6 | 13.6 | 20.7 | 6.3 |
| 2 | P21/c | 40.0 | 46.5 | n.a. | n.a. | 70.9 | n.a. | |
| 2 | P21/c DFT | 37.6 | 45.2 | 39.0 | 39.6 | 7.0 | 63.9 | 39.0 |
3.4 Cambridge Structural Database analysis
These two Cipso–P–M–P torsion angles (φ-angles) are also what we consider to be the best descriptors for the different conformations of the cis-M(PPh3)2-fragment when we searched the CSD to see which of these two conformations was the most common, or if there were indeed other conformations to consider as well. Torsion angles were selected for all cis-bis-triphenylphosphine fragments (P–M–P angles 90–105°), first for all the compounds, giving 1428 structures, and then with the restriction that the metal should be 6-coordinated just as the title compound, leaving us with 287 hits. The data are displayed in Fig. 6.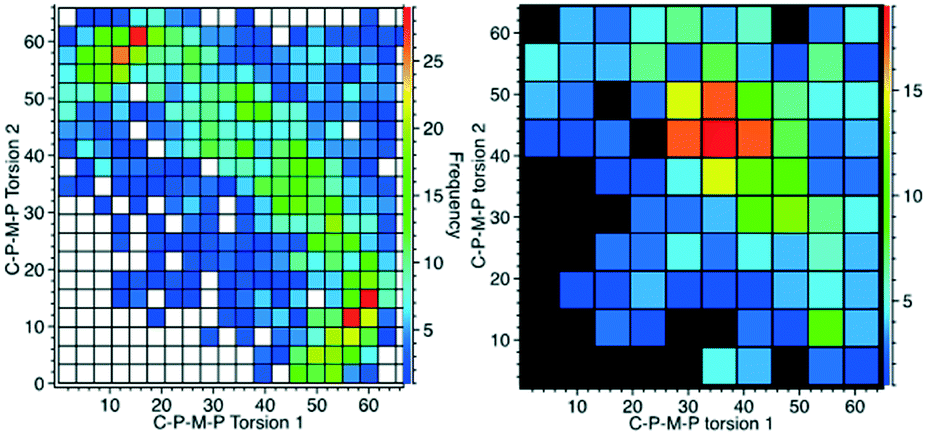 | ||
| Fig. 6 CSD analysis of the Cipso–P–M–P torsion angles for the cis-M(PPh3)2-fragment. Left: all data; right: with the restriction that M should be hexa-coordinated. | ||
What emerges from this is that for lower coordination numbers, as for cyclopentadienyl complexes for example, there is more space for the PPh3 to spread around the metal ion and this seems to generate a clear preference for a 15°/60° conformation. With the more crowded octahedral complexes, like 1 and 2, the most common is instead the 40°/40° conformation. For six-coordinated compounds, there is also a weak tendency towards 20°/20°. This, however, does not tell us anything about the closest phenyl rings exhibiting π-stacking or σ–π-interaction (Fig. 2).
Therefore, we also searched for pairwise C–P–M–P torsion angles less than 65°, as these will correspond to the closest phenyls, and then calculated the angles between the corresponding phenyl planes (β angles). For every angle between planes, there will thus be two torsion angles so two plots are needed to display these data, as shown in Fig. 7. These data show planar π-stacking for the 15°/60° conformation and σ–π-interaction for the 40°/40° conformation. This also corresponds well to the data for both 1 and 2, as can be seen from the β angles also tabulated in Table 3.
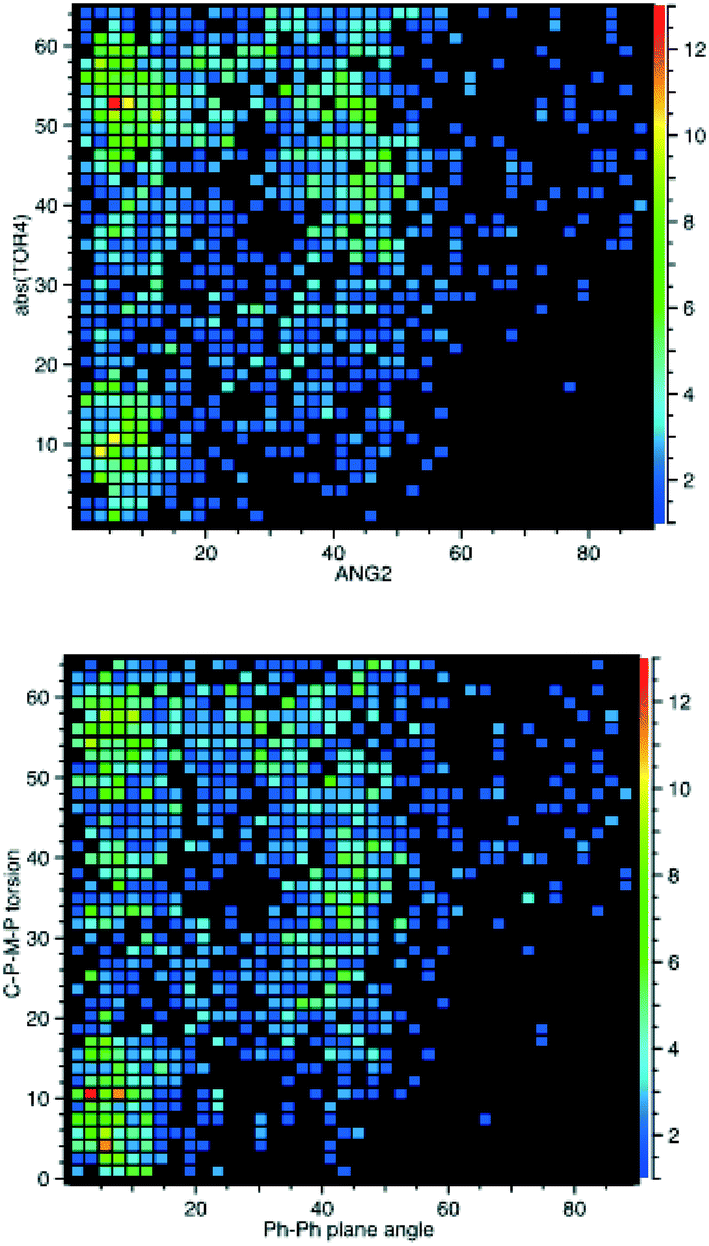 | ||
| Fig. 7 CSD analysis of the Cipso–P–M–P torsion angles for the cis-M(PPh3)2-fragment compared to the angle between the closest phenyl rings of the different PPh3 ligands. | ||
We note that the quantum chemical calculations indicate that the homochiral conformation is preferred. This means that it could be possible to crystallise conglomerates where individual crystals are enantiomerically pure, either ΔΔ or ΛΛ. If this would be the case, these compounds should crystallise in any of the Sohncke space groups (chiral space groups that are without a centre of inversion). However, the most prominent space groups, accounting for 91% of the structures, are the non-Sohncke groups P (#2) and P21/c (#14), and the most occurring “chiral” space group, P212121, is found only in 8 (3%) of the 287 structures.
That both the computational energy minima and distinct peaks in the CSD searcher are found means that the compounds fulfil the criteria for conformational polymorphism, as discussed by Cruz-Cabeza and Bernstein.28
4. Conclusions
Analysis of the CSD data indicates a multitude of possible bis-triphenylphosphine configurations and confirms the notion that both σ–π interactions and π–π stacking are possible.However, quantum chemical calculations show that the heterochiral conformers are substantially higher in energy than the homochiral analogues.
Calculations on the entire crystal indicate, as expected, that the high density phase, 1, is indeed lower in energy than 2, which has a slightly lower density. Hirshfeld surface analysis indicates that a fairly strong double CO⋯H interaction may be one factor giving 1 the lower energy despite the fact that the conformation of the cis-[Mo(CO)4(PPh3)2] molecule in this structure is 12.3 kJ mol−1 more strained than in 2.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
NT, GPM, NK and LÖ thank the Swedish Research Council for a Swedish Research Links grant. SB thanks UCT for the ICRP award. LÖ thanks Ambassade de France and Institut français in Stockholm for a travel grant. We acknowledge access to HPC platforms provided by a GENCI grant (A0030807069).Notes and references
- F. A. Cotton, D. J. Darensbourg, S. Klein and B. W. S. Kolthammer, Inorg. Chem., 1982, 21, 1651–1655 CrossRef.
- W. Reppe and W. J. Schweckendiek, Comp. Rendus, 1948, 560, 104–116 Search PubMed.
- J. A. Osborn, F. H. Jardine, J. F. Young and G. Wilkinson, J. Chem. Soc. A, 1966, 1711–1732 RSC.
- R. Franke, D. Selent and A. Borner, Chem. Rev., 2012, 112, 5675–5732 CrossRef PubMed.
- Phosphorus(III) Ligands in Homogeneous Catalysis, ed. P. C. J. Kamer and P. W. N. M. van Leeuwen, John Wiley & Sons, Ltd., Chichester, U. K., 2012 Search PubMed.
- A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176–4211 CrossRef PubMed.
- H. Zhao, M. Z. Cheng, J. T. Zhang and M. Z. Cai, Green Chem., 2014, 16, 2515–2522 RSC.
- L. A. Aronica, G. Albano, L. Giannotti and E. Meucci, Eur. J. Org. Chem., 2017, 955–963, DOI:10.1002/ejoc.201601392.
- K. Chakrabarti, B. Paul, M. Maji, B. C. Roy, S. Shee and S. Kundu, Org. Biomol. Chem., 2016, 14, 10988–10997 RSC.
- C. Belger and B. Plietker, Chem. Commun., 2012, 48, 5419–5421 RSC.
- J. X. Zheng, L. C. M. Castro, T. Roisnel, C. Darcel and J. B. Sortais, Inorg. Chem. Acta, 2012, 380, 301–307 CrossRef.
- G. Halbritter, F. Knoch, A. Wolski and H. Kisch, Angew. Chem., Int. Ed. Engl., 1994, 33, 1603–1605 CrossRef.
- C. Sauer, Y. Liu, A. De Nisi, S. Protti, M. Fagnoni and M. Bandini, ChemCatChem, 2017, 9, 4456–4459 CrossRef.
- Z. M. Wang, X. Z. Xu and O. Kwon, Chem. Soc. Rev., 2014, 43, 2927–2940 RSC.
- H. M. Liu, Q. M. Zhang, L. M. Wang and X. F. Tong, Chem. Commun., 2010, 46, 312–314 RSC.
- J. M. Brown and K. Mertis, J. Organomet. Chem., 1973, 47, C5–C7 CrossRef.
- V. G. Albano, P. Bellon and M. Sansoni, J. Chem. Soc. A, 1971, 2420–2425, 10.1039/j19710002420.
- J. F. Costello, S. G. Davies, E. T. F. Gould and J. E. Thomson, Dalton Trans., 2015, 44, 5451–5466 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- R. Dovesi, R. Orlando, A. Erba, C. M. Zicovich-Wilson, B. Civalleri, S. Casassa, L. Maschio, M. Ferrabone, M. De La Pierre, P. D'Arco, Y. Noël, M. Causà, M. Rérat and B. Kirtman, Int. J. Quantum Chem., 2014, 114, 1287–1317 CrossRef.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed.
- M. F. Peintinger, D. V. Oliveira and T. Bredow, J. Comput. Chem., 2013, 34, 451–459 CrossRef PubMed.
- F. Cora, A. Patel, N. M. Harrison, C. Roetti, C. Richard and A. Catlow, J. Mater. Chem., 1997, 7, 959–967 RSC.
- B. Nicholls and M. C. Whiting, J. Chem. Soc., 1959, 551–556 RSC.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, D. Jayatilaka and M. A. Spackman, CrystalExplorer, University of Western Australia, Perth, Australia, 2007, http://crystalexplorer.scb.uwa.edu.au Search PubMed.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627–668 CrossRef PubMed.
- A. J. Cruz-Cabeza and J. Bernstein, Chem. Rev., 2014, 114, 2170–2191 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Figure S1. CCDC 1499384 contains the supplementary crystallographic data for compound 2. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ce00337h |
| ‡ Current address: Anorganische Chemie I, Fachrichtung Chemie und Lebensmittelchemie, Technische Universität Dresden, Bergstraße 66, 01062 Dresden, Germany. |
| This journal is © The Royal Society of Chemistry 2018 |