DOI:
10.1039/C8CE00110C
(Paper)
CrystEngComm, 2018,
20, 2812-2821
Pb⋯X (X = N, S, I) tetrel bonding interactions in Pb(II) complexes: X-ray characterization, Hirshfeld surfaces and DFT calculations†
Received
24th January 2018
, Accepted 3rd April 2018
First published on 3rd April 2018
Abstract
Four new Pb(II) complexes of nicotinoylhydrazone and picolinoylhydrazone-based ligands and three different anionic co-ligands (acetate, thiocyanate and iodide) have been synthesized and characterized by structural, analytical and spectroscopic methods. The ligands coordinate to the Pb(II) metal center in a tridentate fashion via two nitrogen and one oxygen donor atoms either in mono-deprotonated or in neutral forms. Single-crystal X-ray crystallography reveals that the molecular complexes aggregate into larger entities depending upon the anion coordinated to the metal centre. The Pb(II) center is hemidirectionally coordinated and, consequently, it is sterically ideal for establishing tetrel bonding interactions. Consequently, in the crystal structures of all the complexes, the Pb participates in short contacts with nitrogen, iodide or sulphur atoms. These contacts are shorter than the sums of the van der Waals radii and larger than the sums of the covalent radii, therefore they can be defined as non-covalent tetrel bonding interactions. They interconnect the covalently bonded units (monomers or dimers) into supramolecular assemblies (1D infinite chains and 3D structures). Hirshfeld surface analysis and fingerprint plots have been used to analyse the contribution of contacts involving the Pb atom. We have analysed the interesting supramolecular assemblies observed in the solid state of all four complexes by means of DFT calculations and characterized them using Bader's theory of atoms-in-molecules.
1. Introduction
The investigation of metal–organic frameworks (MOFs) is continuously increasing the interest of chemists working in solid-state chemistry, crystal engineering and material chemistry because of their promising applications in multidisciplinary fields like photoluminescence and electronics, magnetism, gas absorption, and many others.1 The properties of MOFs can be adjusted by the utilization of different ligands,2 metal centres with different coordination characteristics,3 and counter ions.4 Notably, it is possible to modulate their properties using supramolecular interactions.5
Obviously, hydrogen bonding interactions are the most commonly used force for the construction and/or modification of MOFs. However, other specific non-covalent interactions like highly directional halogen bonding interactions are becoming prominent players in this field.6 In addition to halogen atoms, electron-deficient regions are also present in covalently bonded heavier atoms of groups IV to VII, which are located at the extension of the covalent bond.7 The size and positive electrostatic potential at the σ-holes increase with the increasing electron-withdrawing nature of the covalently bonded group and with the polarizability of the atom on which the σ-hole is formed. Therefore, the strongest σ-hole interactions occur in those complexes where a heavy atom of groups IV to VII is covalently bonded to electronegative ones. This type of σ-hole interaction has been widely described for groups IV to VII (chalcogen, pnicogen and halogen bonding interactions), however those of group IV (tetrel interactions) are less common,8 It has been shown that they can be markedly strong, in particular with Sn and Pb, the metallic members of the group. In this regard, we have reported the design and synthesis of lead(II) supramolecular metal–organic frameworks (SMOFs) based on covalent and non-covalent tetrel bonding interactions.9 In the solid state, non-covalent tetrel bonds interconnect the covalently bonded units into supramolecular assemblies. A combined DFT study and a statistical survey of the Cambridge Structural Database (CSD) revealed that tetrel bonds with a hemidirectionally coordinated lead center occur with high probability and with predictable geometries, thus playing an important role in the solid-state chemistry of lead.9a Moreover, we have also shown that concurrent non-covalent tetrel bonding, agostic interactions and chelate ring–chelate ring stacking interactions control the supramolecular architectures and organometallic frameworks observed in the solid state architecture of hemidirectionally coordinated PbX2 salts (X = Cl, NO2, I and SCN) to the N′-(phenyl(pyridin-2-yl)methylene)-isonicotinohydrazide ligand.10
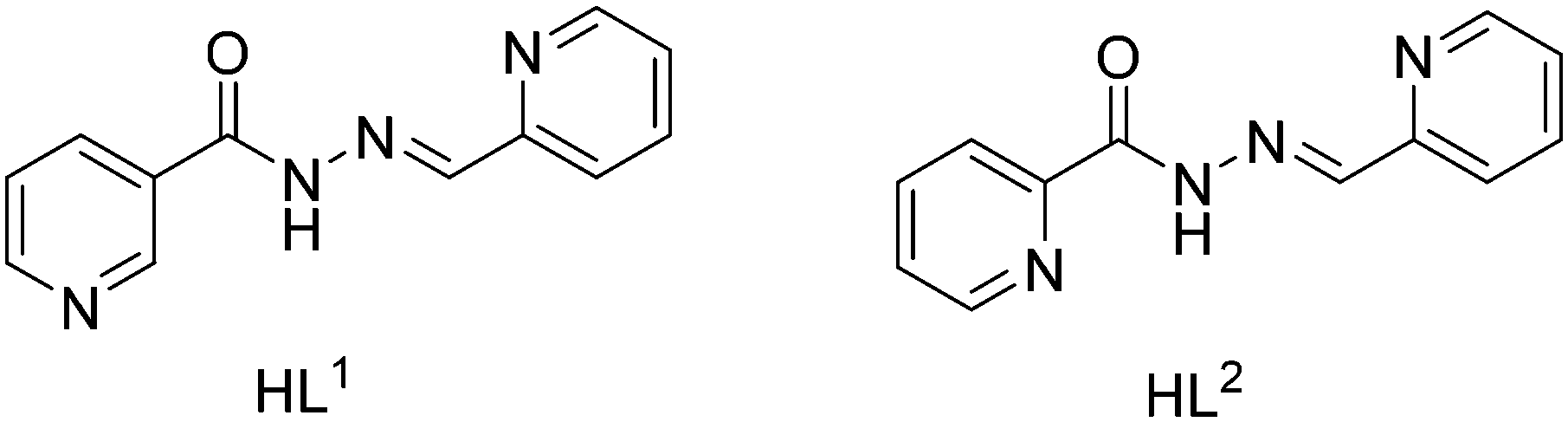
Taking advantage of this understanding, herein we report the synthesis and X-ray characterization of four new Pb(II) complexes with nicotinoylhydrazone and picolinoylhydrazone-based ligands and different anions (acetate, thiocyanate and iodide) as co-ligands (see Scheme 1). Interestingly, in all complexes, Pb(II) presents a hemidirectional coordination pattern, thus presenting a clear void in the distribution of the bonds to the ligands. This facilitates the approach of electron donors and their participation in the non-covalent tetrel bonding interactions. These forces along with hydrogen bonding and π-stacking interactions control the supramolecular architectures observed in their solid state architecture. The nature of the tetrel bonds in all the four structures was studied by DFT calculations and molecular electrostatic potential (MEP) calculations, which showed the presence of the σ-hole in the Pb atom and the considerable strength of these interactions.
 |
| | Scheme 1 Ligands used in this work. | |
2. Experimental
2.1 Syntheses
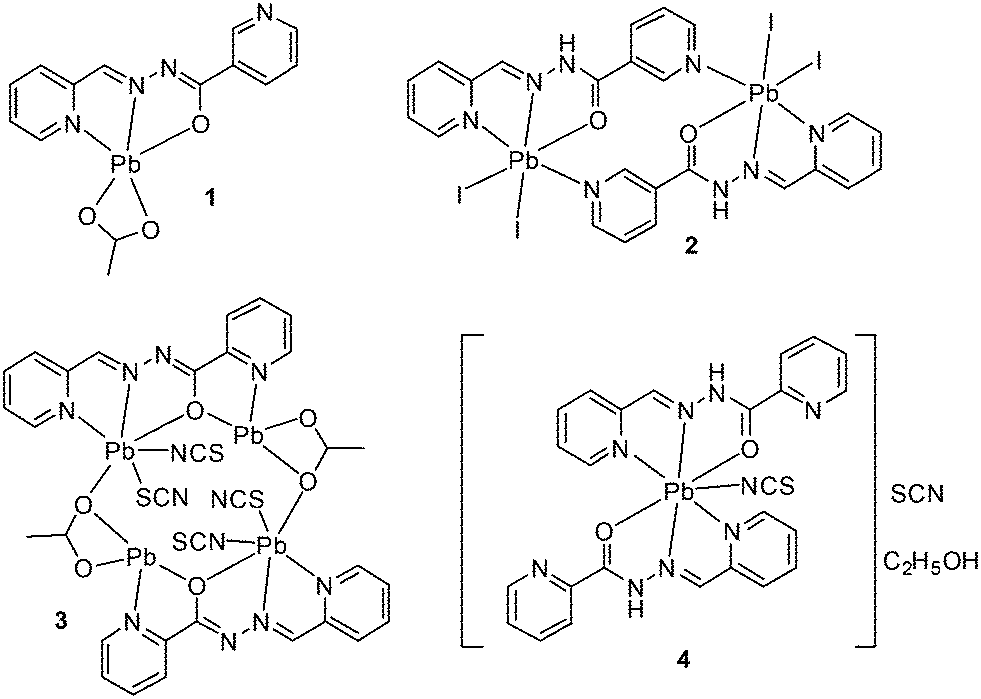
The schematic presentation of the ligands used is depicted in Scheme 1. Due to the insolubility of these compounds in most of the common solvents employed, we failed to crystallize the materials as single crystals, rather than polycrystalline powders. A possible future solution to our inability to grow single-crystals is the use of very interesting and unusual glassware for the reaction/crystallization apparatus (branched tube, see Scheme S1†) which was recently developed by us,11 and that the syntheses were performed in it. The detailed synthesis of complex (1) is described here. The rest of the complexes were synthesized using the same procedure, mixing equimolar quantities of the appropriate PbX2 salts (X = CH3COO, I, with HL1 and X = CH3COO, SCN with HL2).
Synthesis of [Pb(L1)(CH3COO)] (1).
Pb(CH3COO)2 and HL1 (0.164 g, 0.5 mmol; 0.113 g, 0.5 mmol) were placed in the main arm of the branched tube. Methanol or ethanol (15 ml) was carefully added to fill the arms. The tube was sealed and immersed in an oil bath at 60 °C while the branched arm was kept at ambient temperature (see Scheme S1†). After 4 days, crystals of (1) that were isolated in the cooler arm were filtered off, washed with acetone and ether, and dried in air.
Synthesis of 2–3.
These compounds were prepared in the same way as 1. To prepare 2, we used PbI2, and HL1 (0.230 g, 0.5 mmol; 0.113 g, 0.5 mmol) and methanol (15 ml) as the solvent. For 3, Pb(CH3COO)2 (0.164 g, 0.5 mmol) and KSCN (0.097 g, 1.0 mmol) with HL2 (0.113 g, 0.5 mmol) and ethanol (15 ml) as the solvent were used. For 4, we used Pb(SCN)2 and HL2 (0.162 g, 0.5 mmol; 0.113 g, 0.5 mmol) and ethanol (15 ml) as the solvent. For more information, see our published paper.11
[Pb(L1)(CH3COO)] (1).
The isolated yield was 72%. Anal. calcd. (found) for C14H12N4O3Pb; C, 34.21 (34.01); H, 2.46 (2.51); N, 11.40 (11.60)%. IR (cm−1) selected bands: ṽ = CH b (oop): 667 (m) and 779 (m); COOst: 1385 (s); CCst: 1460 (m); C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N st: 1499 and 1579 (m); CH st: 3007 (w) cm−1.
N st: 1499 and 1579 (m); CH st: 3007 (w) cm−1.
[Pb(HL1)(I)2]2 (2).
The isolated yield was 87%. Anal. calcd. (found) for C12H10I2N4OPb; C, 25.72 (25.91); H, 1.80 (1.75); N, 10.00 (10.15)%. IR (cm−1) selected bands: ṽ = CH b (oop): 697 (m) and 769 (m); CCst: 1436 (m); CN st: 1529 and 1589 (m); C–O st (ligand) 1635; CH st: 2925 (w) cm−1.
[Pb2(L2)(CH3COO)(SCN)2]2 (3).
The isolated yield was 85%. Anal. calcd. (found) for C16H12N6O3Pb2S2; C, 23.58 (23.61); H, 1.48 (1.58); N, 10.31 (10.42)%. IR (cm−1) selected bands: ṽ = CH b (oop): 632 (m) and 749 (m); COOst: 1352 (s); CCst: 1463 (m); CN st: 1506 and 1561 (m); SCN: 2012 and 2055; CH st: 2855 (w) cm−1.
[Pb(HL2)2(SCN)2](C2H5OH) (4).
The isolated yield was 85%. Anal. calcd. (found) for C28H26N10O3PbS2; C, 40.92 (40.71); H, 3.19 (3.28); N, 17.04 (17.22)%. IR (cm−1) selected bands: ṽ = CH b (oop): 685 (m) and 742 (m); CCst: 1466 (m); CN st: 1525 and 1580 (m); CO st (ligand) 1650; SCN: 2036 and 2067; CH st: 2926 (w); NH: 3213 (m) cm−1.
2.2 Crystallographic analyses
Single crystals of (1–4, see Scheme 2) suitable for X-ray analysis were selected and crystallographic data were collected on an Xcalibur 2 for (1), a Bruker-AXS Kappa Apex II for (2), a SuperNova (Dual, Cu at zero, Atlas) (3), or a Bruker APEX-II CCD (4) diffractometer using MoKα radiation (λ = 0.71073 Å) in the ω-scan mode. The detector frames were integrated by using the program SAINT,12 and the empirical absorption corrections were performed using the SADABS program.13 All the structures were solved by direct methods and refined by the full matrix least-squares procedures in SHELXTL.14 All non-hydrogen atoms were refined with anisotropic displacement parameters, whereas the hydrogen atoms were placed in calculated positions, when possible, and given isotropic U values that are 1.2 times that of the atom to which they are bonded. The materials for publication were prepared using the SHELXTL,14 PLATON15 and DIAMOND 3.1 (ref. 16) programs. Further details regarding data collection and structure refinement are given in Table 1. The structure of 4 exhibits large displacement ellipsoids in the solvent (the ethanol molecule) and to a lesser degree in the anions. The attempts to model these as structural disorder failed to provide more reasonable structural models. The positions of the hydrogen atoms bonded to the nitrogen could not be satisfactorily refined, and were placed on calculated positions.
 |
| | Scheme 2 Compounds 1–4 synthesized in this work. | |
Table 1 Crystal data and structure refinement parameters for (1–4)
| Structure |
1
|
2
|
3
|
4
|
| Empirical formula |
C28H24N8O6Pb2 |
C12H10I2N4O1Pb1 |
C16H12N6O3Pb2S2 |
C28H26N10O3Pb1S2 |
| Formula weight |
982.95 |
687.23 |
814.82 |
821.90 |
| Temperature (K) |
100(2) |
170(2) |
130.0(1) |
293(2) |
| Wavelength (Å) |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
| Crystal system |
Monoclinic |
Triclinic |
Triclinic |
Monoclinic |
| Space group |
P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21/c |
|
a, b, c (Å) |
7.4785(2), 21.4405(5), 9.0220(2) |
6.674(6), 7.898(7), 16.7940(15) |
8.5997(9), 10.9376(7), 11.7938(12) |
15.4161(5), 13.7171(5), 14.9481(5) |
|
α, β, γ (°) |
90, 94.218(2), 90 |
82.614(5), 81.440(5), 65.269(5) |
72.684(7), 80.451(9), 82.758(7) |
90, 94.944(3), 90 |
| Volume (Å3) |
1442.69(6) |
792.99(13) |
1040.90(17) |
3149.23(19) |
|
Z/density (calc.) (Mg m−3) |
2/2.263 |
2/2.878 |
2/2.600 |
4/1.733 |
| Absorption coefficient (mm−1) |
11.713 |
14.53 |
16.384 |
5.537 |
|
F(000) |
920 |
612 |
740 |
1608 |
| Crystal size (mm3) |
0.33 × 0.17 × 0.09 |
0.089 × 0.056 × 0.051 |
0.3829 × 0.1692 × 0.1349 |
0.41 × 0.27 × 0.19 |
|
θ range for data collection |
2.89 to 31.00 |
2.46 to 26.99 |
2.978 to 26.998 |
3.935 to 26.998 |
| Limiting indices |
−10 ≤ h ≤ 10 |
−8 ≤ h ≤ 8 |
−10 ≤ h ≤ 10 |
−15 ≤ h ≤ 19 |
| −31 ≤ k ≤ 30 |
−10 ≤ k ≤ 9 |
−13 ≤ k ≤ 13 |
−10 ≤ k ≤ 17 |
| −12 ≤ l ≤ 13 |
−21 ≤ l ≤ 21 |
−15 ≤ l ≤ 15 |
−19 ≤ l ≤ 19 |
| Completeness to θ (%) |
99.7 |
98.8 |
99.9 |
98.2 |
| Absorption correction |
Semi-empirical from equivalents |
Semi-empirical from equivalents |
Semi-empirical from equivalents |
Semi-empirical from equivalents |
| Max. and min. transmission |
0.36 and 0.09 |
0.534 and 0.746 |
0.170 and 0.034 |
— |
| Refinement method |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
| Data/parameters |
4582/203 |
3413/182 |
4469/263 |
6750/399 |
| Goodness-of-fit on F2 |
1.043 |
1.11 |
1.042 |
0.845 |
| Final R indices [I > 2σ(I)] |
R
1 = 0.018, wR2 = 0.042 |
R
1 = 0.0897, wR2 = 0.2154 |
R
1 = 0.0249, wR2 = 0.0555 |
R
1 = 0.0291, wR2 = 0.0559 |
|
R indices (all data) |
R
1 = 0.0226, wR2 = 0.043 |
R
1 = 0.1228, wR2 = 0.2394 |
R
1 = 0.0285, wR2 = 0.0574 |
R
1 = 0.05660, wR2 = 0.0580 |
| Largest diff. peak and hole (e Å−3) |
1.719 and −0.586 |
10.83 and −1.99 |
1.552 and −1.936 |
0.783 and −0.621 |
| CCDC no |
1588614
|
1588615
|
1588616
|
1588617
|
2.3 Hirshfeld surface analysis
Molecular Hirshfeld surfaces17–19 in the crystal structure are constructed based on the electron distribution calculated as the sum of spherical atom electron densities.20 For a given crystal structure and set of spherical atomic electron densities, the Hirshfeld surface is unique.21 The normalized contact distance (dnorm) based on de (distance from the point to the nearest nucleus external to the surface), di (distance to the nearest nucleus internal to the surface) and the vdW radii of the atom, given by eqn (1) enables the identification of the regions of particular importance to the intermolecular interactions.17 The combination of de and di in the form of a 2D fingerprint plot22 provides a summary of the intermolecular contacts in the crystal.17 The Hirshfeld surfaces are mapped using dnorm, and the 2D fingerprint plots presented in this paper were generated using CrystalExplorer 3.1.23| |  | (1) |
2.4 Theoretical methods
The geometries of the complexes included in this study were computed at the M06-2X/def2-TZVP level of theory using the crystallographic coordinates within the TURBOMOLE program.24 The advantage of the M06-2X functional is the correct description of the intermediate-range van der Waals interaction, due to the inclusion of the kinetic energy density, which helps to identify weak non-covalent bonds as those reported herein. The basis set superposition error for the calculation of interaction energies has been corrected using the counterpoise method.25 The “atoms-in-molecules” (AIM)26 analysis of the electron density has been performed at the same level of theory using the AIMAll program.27
3. Results and discussion
Structural description of 1–4 focusing on the tetrel bonding interactions
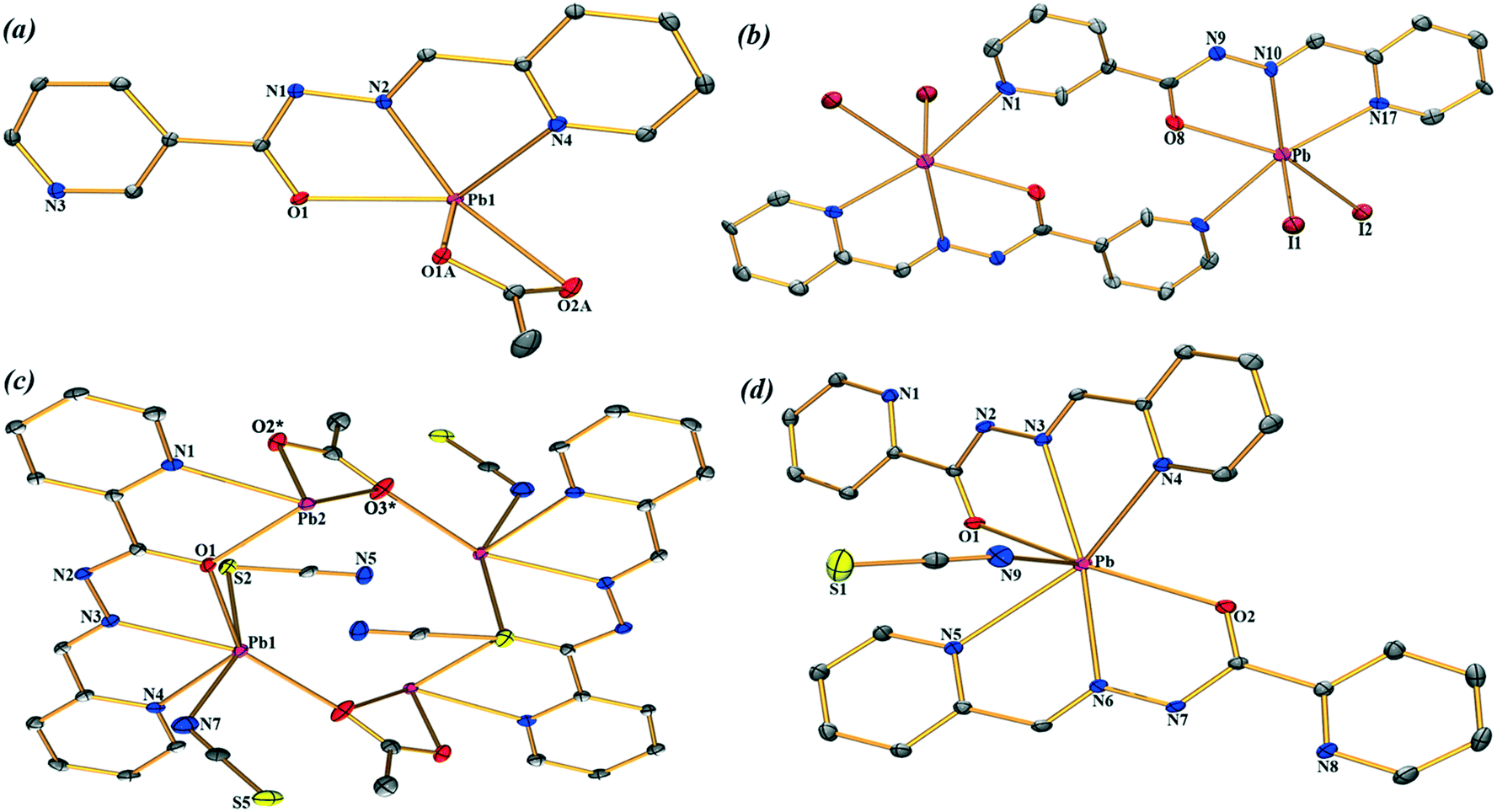
The molecular views of the asymmetric unit of the title Pb compounds (1–4) are depicted in Fig. 1 with an atom numbering scheme. These structures exhibit hydrogen bonds and π–π stacking interactions (Tables S1 and S2†). The self-assemblies of the title structures are described in detail in the ESI.† We are particularly interested in exploring the less studied tetrel bonding interactions. These interactions are present in the four structures reported herein and are important in the formation of supramolecular assemblies in their solid state.
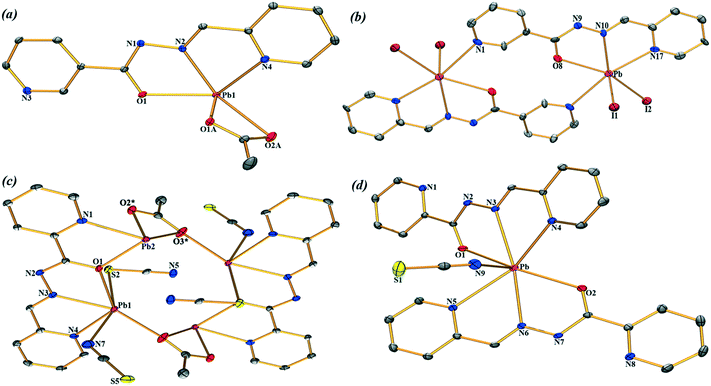 |
| | Fig. 1 Central atom environments (ORTEP drawing) with an atom numbering scheme of (1–4) (a–d respectively). The solvent molecules and hydrogen atoms are omitted for clarity. For compound (3), the atoms labelled with * are generated at the equivalent position (−x, −y, 1 − z). The thermal ellipsoids are drawn at the 25% probability level. | |
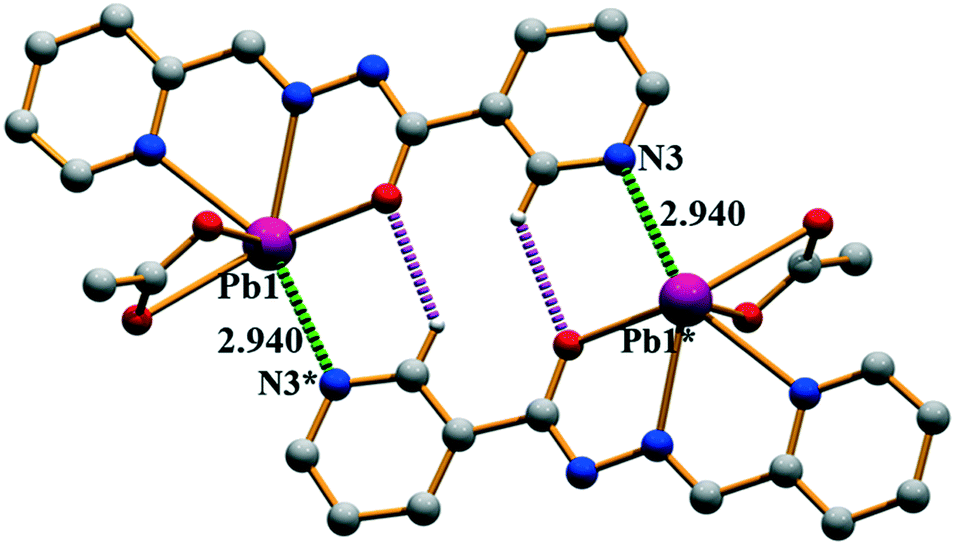
Compound 1 crystallizes in the P21/c space group where Pb(II) is five-coordinated, in a distorted tetragonal-pyramid fashion, by the O1, N2 and N4 atoms of the ligands and both the O atoms of the acetate co-ligand. A two-dimensional (2D) assembly in the (101) plane is generated through the combination of two types of C–H⋯O hydrogen bonds and Pb⋯N tetrel bonds, as detailed in Fig. S1a (see the ESI†). In Fig. 2, we represent a self-assembled dimer extracted from the 2D plane that is characterized by the presence of two symmetrically equivalent Pb⋯N tetrel bonds [2.940(2) Å]. The Pb⋯N distance is longer than the sum of the covalent radii (2.17 Å) and shorter than the sum of the van der Waals radii (∑RvdW = 3.57 Å), thus confirming the non-covalent nature of this interaction that is further analysed below.
 |
| | Fig. 2 Self-assembled dimer generated through Pb⋯N tetrel bonds and C–H⋯O hydrogen bonds in (1). Distances are in Å. The symmetry operation used to generate equivalent atoms (* = −x, −y, 1 − z). | |
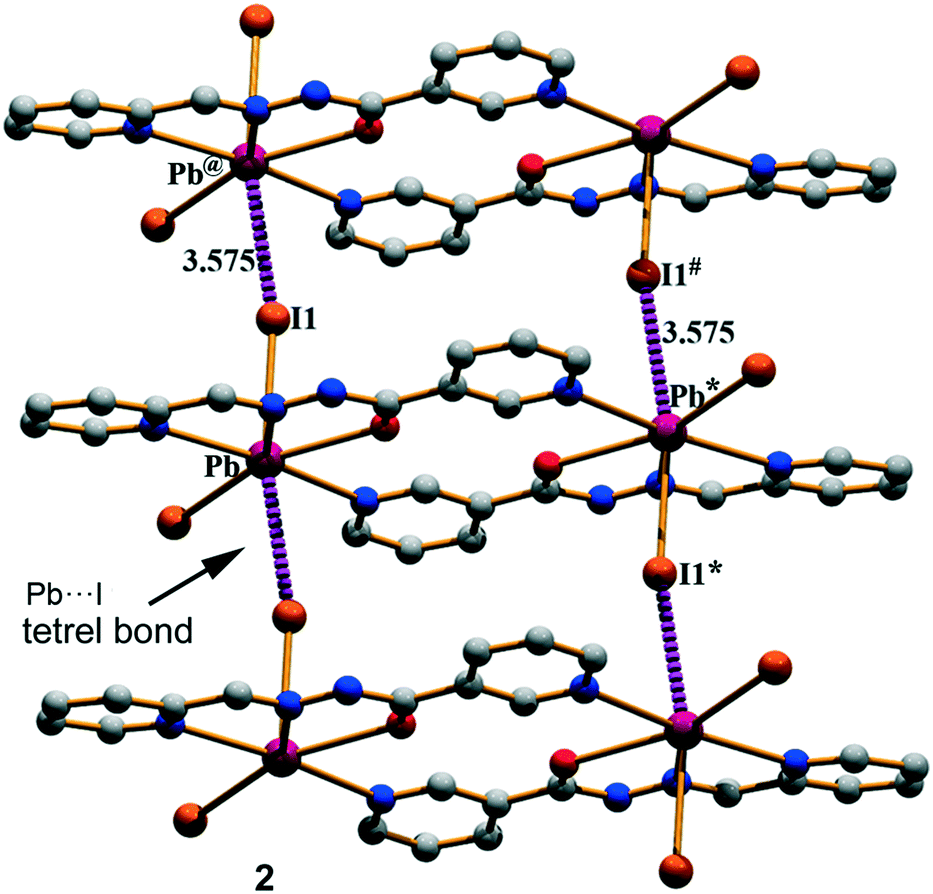
Compound 2 has a crystallographic inversion center at the midpoint and thus it contains two symmetry related [PbHL1I2] units. In the solid state of this compound, infinite 1D columns, along the [100] direction, are formed, which are governed by the formation of I⋯Pb tetrel bonds with a separation distance of 3.575(4) Å. This distance is considerably shorter than ∑RvdW = 4.00 Å and also much longer that the sum of the covalent radii (2.85 Å), thus confirming the non-covalent nature of the interaction (see Fig. 3). The solid-state structure of 2 is further stabilized through a combination of N–H⋯I, C–H⋯O, and C–H⋯I bonds and π–π stacking interactions as detailed in Fig. S2 and Tables S1 and S2 in the ESI.†
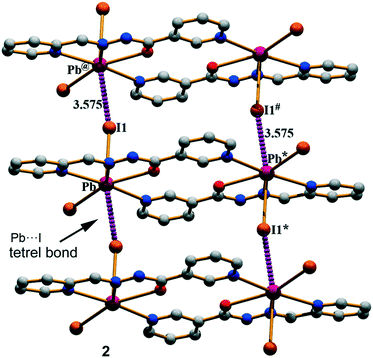 |
| | Fig. 3 Perspective view of the infinite 1D column generated through Pb⋯I tetrel bonds in (2). Distances are in Å. The symmetry operation used to generate equivalent atoms (* = −x, 1 − y, 2 − z; # = 1 − x, 1 − y, 2 − z; @ = 1 + x, y, z). | |
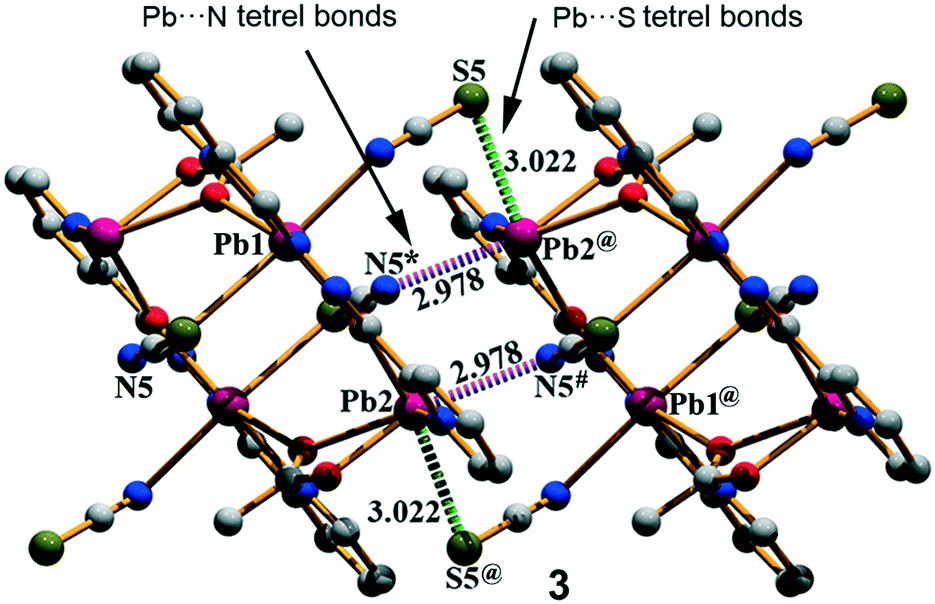
Complex 3 crystallizes in the triclinic space group P and features a centrosymmetric acetate bridged tetranuclear Pb moiety. The Schiff base ligand coordinates two Pb ions (Pb1 and Pb2) and the acetate oxygen atoms O2 and O3. The tetranuclear molecular moiety is generated by the acetate bridges. Interestingly, in compound 3, two different tetrel bonding interactions are established (see Fig. 4) in the solid state, thus confirming the ability of the Pb(II) atom to participate in σ-hole interactions. One tetrel bond is formed between the S(5) atom of the N-coordinated pseudohalide ligand and the Pb(2) atom characterized by an interatomic distance of 3.022(2) Å, which is in between the sum of the covalent and vdW radii (2.51 and 3.82 Å, respectively). The second tetrel bond is established between the N(5) atom of the S-coordinated pseudohalide anion and the Pb(2) metal center with an interatomic distance of 2.978(4) Å, which is considerably longer than the sum of the covalent radii (2.17 Å) and slightly shorter than the sum of the van der Waals radii (∑RvdW = 3.57 Å). Both tetrel bonding interactions are responsible for the formation of the self-assembled dimer shown in Fig. 4. Moreover, the solid-state structure of compound 3 also exhibits C–H⋯S and C–H⋯O bonds, and π–π stacking that generate 2D layers as further discussed in the ESI.†
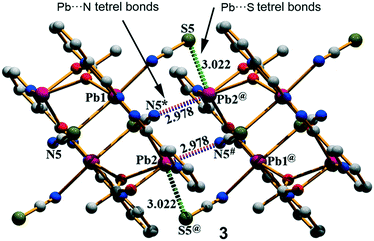 |
| | Fig. 4 The self-assembled dimer generated through Pb⋯N tetrel bonds in (3). Distances are in Å. The symmetry operation used to generate equivalent atoms (* = −x, −y, 1 − z; # = 1 + x, y, z; @ = 1 − x, −y, 1 − z). | |
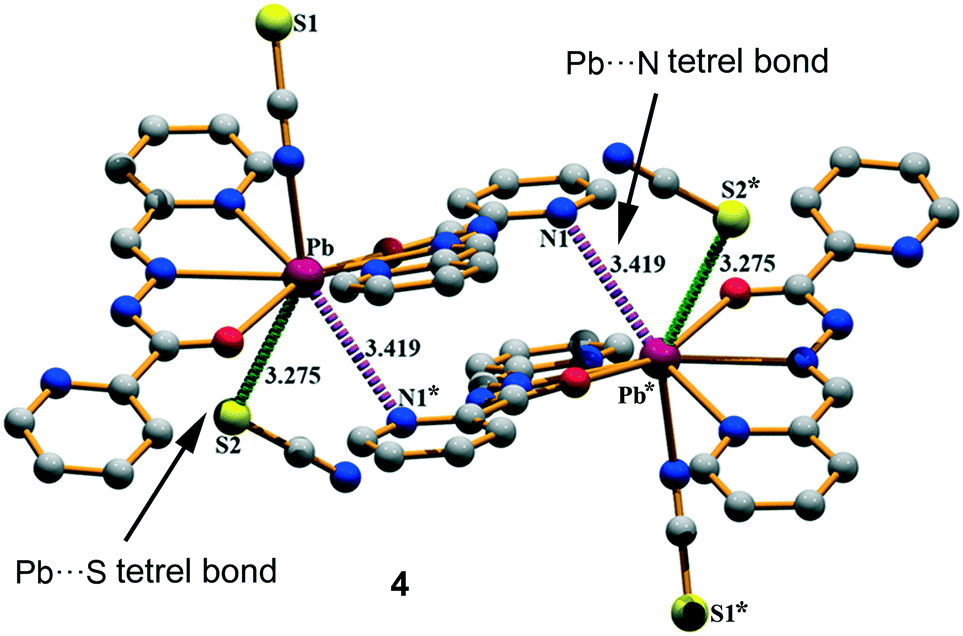
Finally, compound 4 crystallizes in the monoclinic P21/c space group with a hepta-coordination of the Pb(II) metal center (see Fig. 1d). This compound also exhibits two types of tetrel bonding interactions in the solid state as depicted in Fig. 5. One involves the non-coordinated counter-anion (SCN−) that is located at 3.275(2) Å from the Pb. The second tetrel bond (Pb⋯N interaction, 3.419 Å) is responsible for the formation of the self-assembled dimer shown in Fig. 4 along with the π–π stacking interactions, which is described in Fig. S4 (ESI†).
 |
| | Fig. 5 Self-assembled dimer generated through Pb⋯N/S tetrel bonds in (4). Distances are in Å. The symmetry operation used to generate equivalent atoms (* = 1 − x, 1 − y, 2 − z). | |
Hirshfeld surface analysis
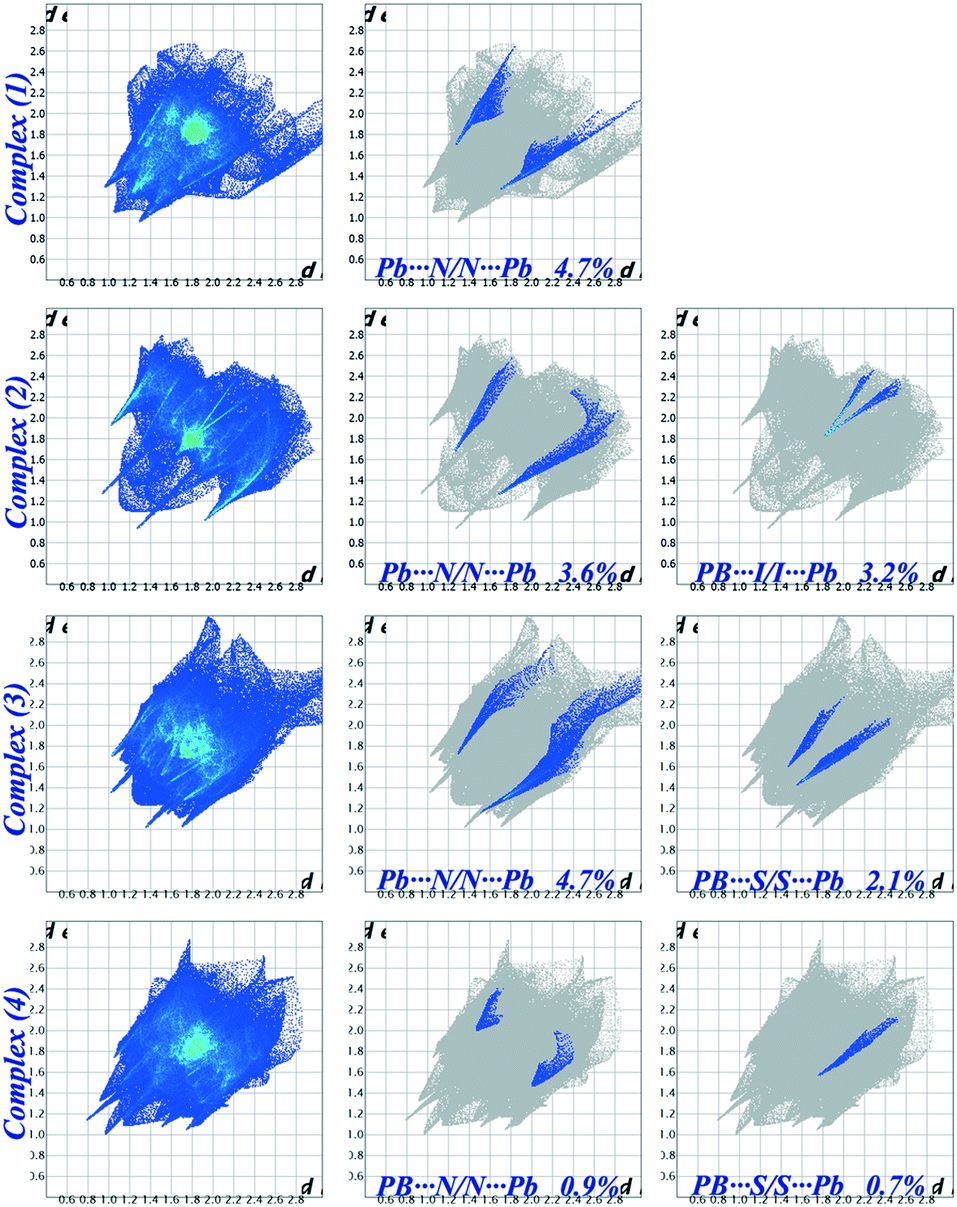
The Hirshfeld surfaces of the title compounds (1–4) are illustrated in Fig. S5 (ESI†), which shows surfaces that have been mapped over dnorm [−0.355 to 1.565 Å in (1); −0.376 to 1.324 Å in (2), −0.523 to 1.369 Å in (3) and −0.533 to 1.236 Å (4)]. The dominant intermolecular interactions are evident by the circular depressions on the dnorm surface of the structures (Fig. S5, ESI†). We are particularly interested to explore the contributions of the (Pb⋯N, Pb⋯I, and Pb⋯S) tetrel bonds that are involved within the structures and appear as spikes in the fingerprint plots (Fig. 6). The interactions in between the Pb and N atoms have a clear signature by the spikes in the donor and acceptor regions of the decomposed fingerprint plot with de + di ≈ 2.963 Å, de + di ≈ 2.942 Å and de + di ≈ 3.448 Å for (1), (2) and (4) respectively (Fig. 3). In complex (3), the Pb⋯N interaction has been evidenced in the region de + di ≈ 2.672 Å whereas its N⋯Pb counterpart is located at de + di ≈ 2.987 Å (see the 2nd column of Fig. 6). This is only due to the variation in the Pb⋯N interaction (3.9%) with N⋯Pb (0.8%). The breakdown of the fingerprint plot shows that the Pb⋯N/N⋯Pb interactions comprise only 4.7%, 3.6%, 4.7% and 0.9% of the total Hirshfeld surface of the molecules in (1–4), respectively (Table S3, ESI†). The spikes in the region di + de ≈ 3.602 Å designate the tetrel bonds between the Pb and I atoms in complex (2) (3rd column of Fig. 6). The percentages of contribution of these interactions to the relative Hirshfeld surface areas reveal a noticeable involvement of the Pb⋯I/I⋯Pb interactions (3.2%) out of which their Pb⋯I and I⋯Pb counterparts comprise 1.7% and 1.5% of the surface area in (2). The percentages of contribution corresponding to the Pb⋯S/S⋯Pb interactions in (3) and (4) are very different. The spikes in the regions de + di ≈ 2.981 Å and de + di ≈ 3.042 Å are due to their Pb⋯S and S⋯Pb counterparts which contribute 1.6% and 0.5% to the total Hirshfeld surface area in (3). In complex (4), there is no signature about the S⋯Pb counterpart (0.0% contribution) and the only spike available in the region de + di ≈ 3.312 Å designate the Pb⋯S interaction which contributes 0.7% only to the total Hirshfeld surface area (Fig. S6, ESI†). The decomposed dnorm surfaces have been mapped to identify and to visualize the contribution of tetrel bonds involved within the structures (1–4) (Fig. S6, ESI†).
 |
| | Fig. 6 Fingerprint plots of (1–4): Full (left) and resolved into tetrel bonds [Pb⋯N (middle) and Pb⋯S (right)] showing the percentages of contacts contributing to the total Hirshfeld surface area of the molecules. | |
Theoretical study
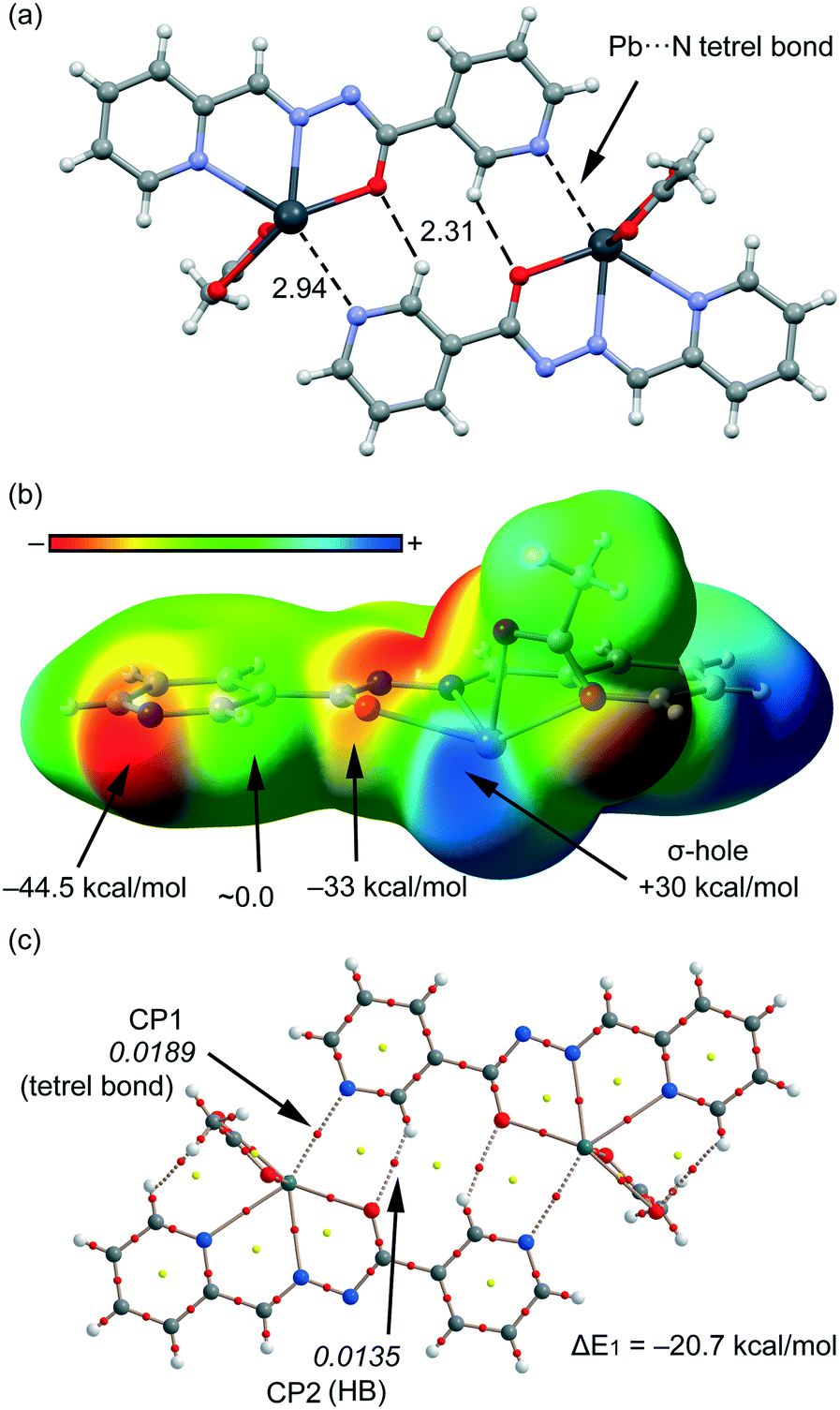
We have focused the theoretical study to the comparison of the energetic features of the different tetrel bonding interactions observed in the crystal packing of compounds 1–4 described above. In Fig. 7a, we represent a dimer found in the X-ray solid-state structure of compound 1 that is formed by a combination of C–H⋯O hydrogen bonds and Pb⋯N tetrel bonding interactions (see black dashed lines). These interactions play a key role in the formation of the supramolecular network highlighted in Fig. S1a.† We have represented in Fig. 7b the MEP surface of compound 1, and the presence of a strong σ-hole (+30 kcal mol−1) in the Pb atom at the extension of the O–Pb bond can be observed. The electrostatic potential at the N atom of the pyridine is negative (−44.5 kcal mol−1), thus indicating that the tetrel bond is electrostatically very favourable. In fact, it is more favourable than H-bonding since the MEP value at the H atom is negligible. We have computed the interaction energy of this dimer (see Fig. 7c), which is large and negative (ΔE1 = −20.7 kcal mol−1) due to the formation of two symmetrically equivalent tetrel bonds and also two C–H⋯O H-bonds. We have used Bader's theory of atoms in molecules26 to characterize the interactions. The existence of a bond critical point (CP) and bond path connecting two atoms is a clear evidence of interaction, since it indicates that the electron density is accumulated between the nuclei that are linked by the associated atomic interaction line.28 In Fig. 7c, we represent the distribution of CPs and bond paths of the dimer and it shows that H-bonds are characterized by a bond CP (red sphere) and a bond path connecting the H atoms to the O atoms (ρBCP = 0.0135 a.u.). Moreover, the Pb⋯N tetrel bonding interaction is characterized by a bond CP and a bond path interconnecting the N and Pb atoms (ρBCP = 0.0189 a.u.). The value of ρ at the bond CP that characterized the tetrel bond is larger than that at the H-bond, indicating that the tetrel bond is stronger, which is in agreement with the MEP analysis.
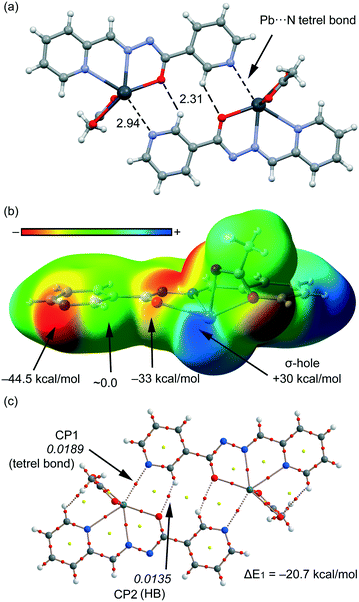 |
| | Fig. 7 (a) Tetrel bonded dimer from the crystal packing of 1. Distances are in Å. (b) MEP surface (isosurface = 0.001 a.u.) of compound 1. MEP values at selected points of the surface are indicated. A theoretical model used to evaluate the binding energy. (c) AIM distribution of bond and ring critical points (red and yellow spheres, respectively) and bond paths for the dimer of 1. The values of ρ(r) at the bond CPs are given in italics. | |
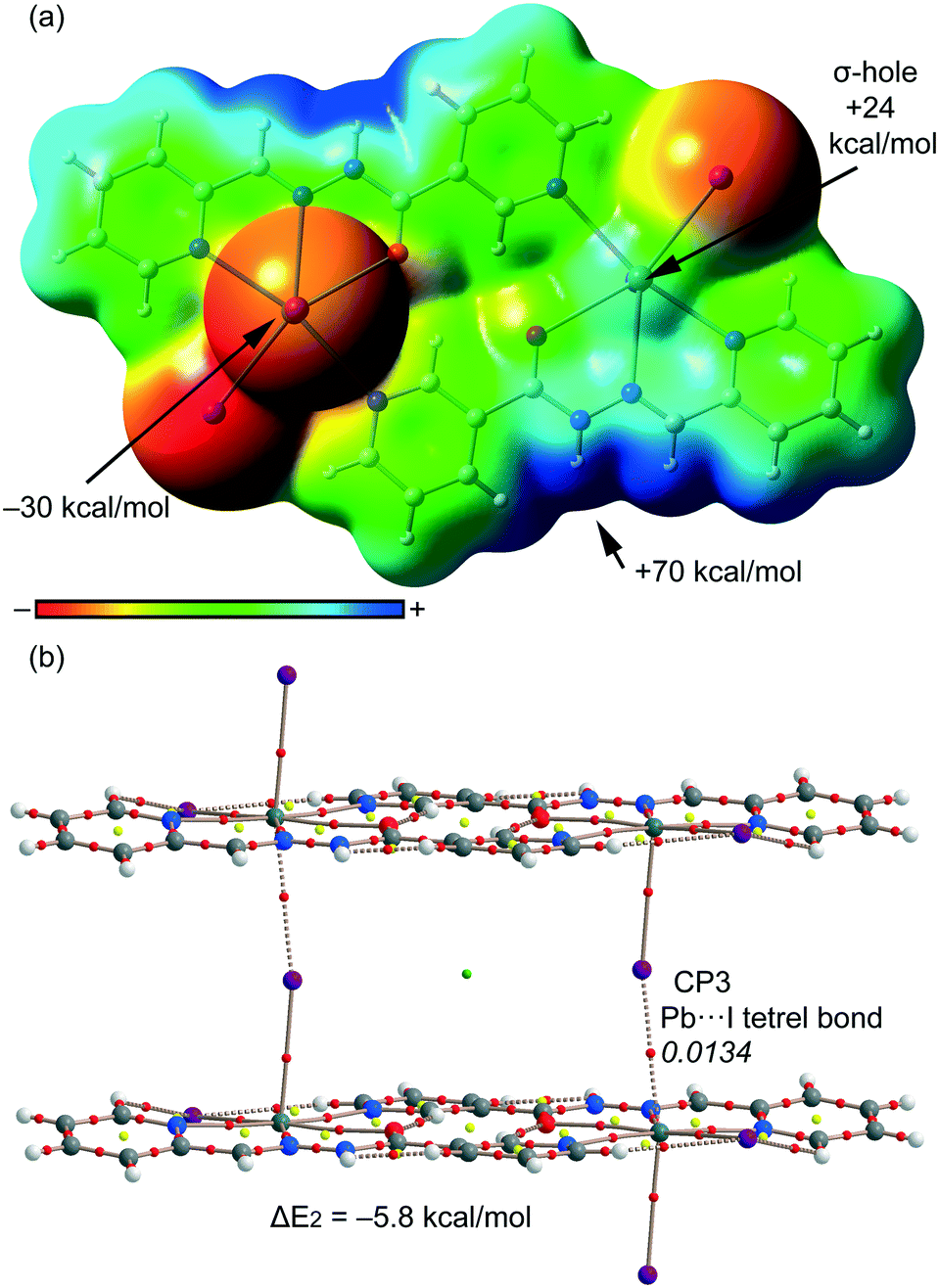
For compound 2, we have studied theoretically the interesting 1D supramolecular polymeric chain (see Fig. 3) that is formed in the solid-state by evaluating the interaction energy of a dimeric model extracted from the X-ray structure. In order to examine the nucleophilic and electrophilic regions in compound 2 and rationalize the donor-acceptor interactions, we have first computed the MEP surface (see Fig. 8a). A σ-hole is also observed in this compound at the extension of the I–Pb bond, however, the MEP value (+24 kcal mol−1) is considerably smaller than that computed for compound 1 (+30 kcal mol−1) thus anticipating a weaker tetrel bonding interaction. Moreover, the MEP value at the I atom is also smaller in terms of absolute value than that at the pyridine N atom in 1. As a consequence, the interaction energy computed for the tetrel bonded complex is modest (ΔE2 = −5.8 kcal mol−1). We have used the AIM analysis and distribution of CPs to confirm the existence of the Pb⋯I tetrel bond (see Fig. 8b). Each σ-hole interaction is characterized by the presence of a bond CP (ρ = 0.0134 a.u.) and a bond path inter-connecting the I and Pb atoms (see Fig. 8b). The value of ρ at the bond CP is smaller in 2 than in 1, which is in agreement with the weaker interaction energy.
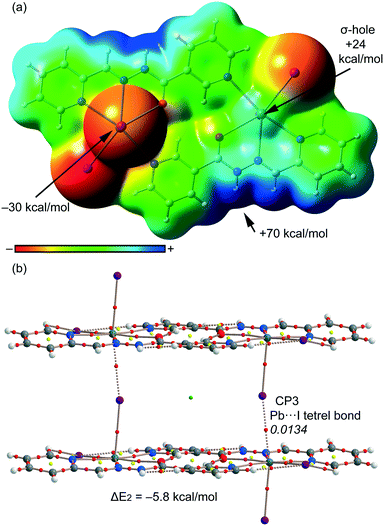 |
| | Fig. 8 (a) MEP surface (isosurface = 0.001 a.u.) of compound 2. MEP values at selected points of the surface are indicated. A theoretical model used to evaluate the binding energy. (b) AIM distribution of bond and ring critical points (red and yellow spheres, respectively) and bond paths for the dimer of 2. The values of ρ(r) at the bond CPs are given in italics. | |
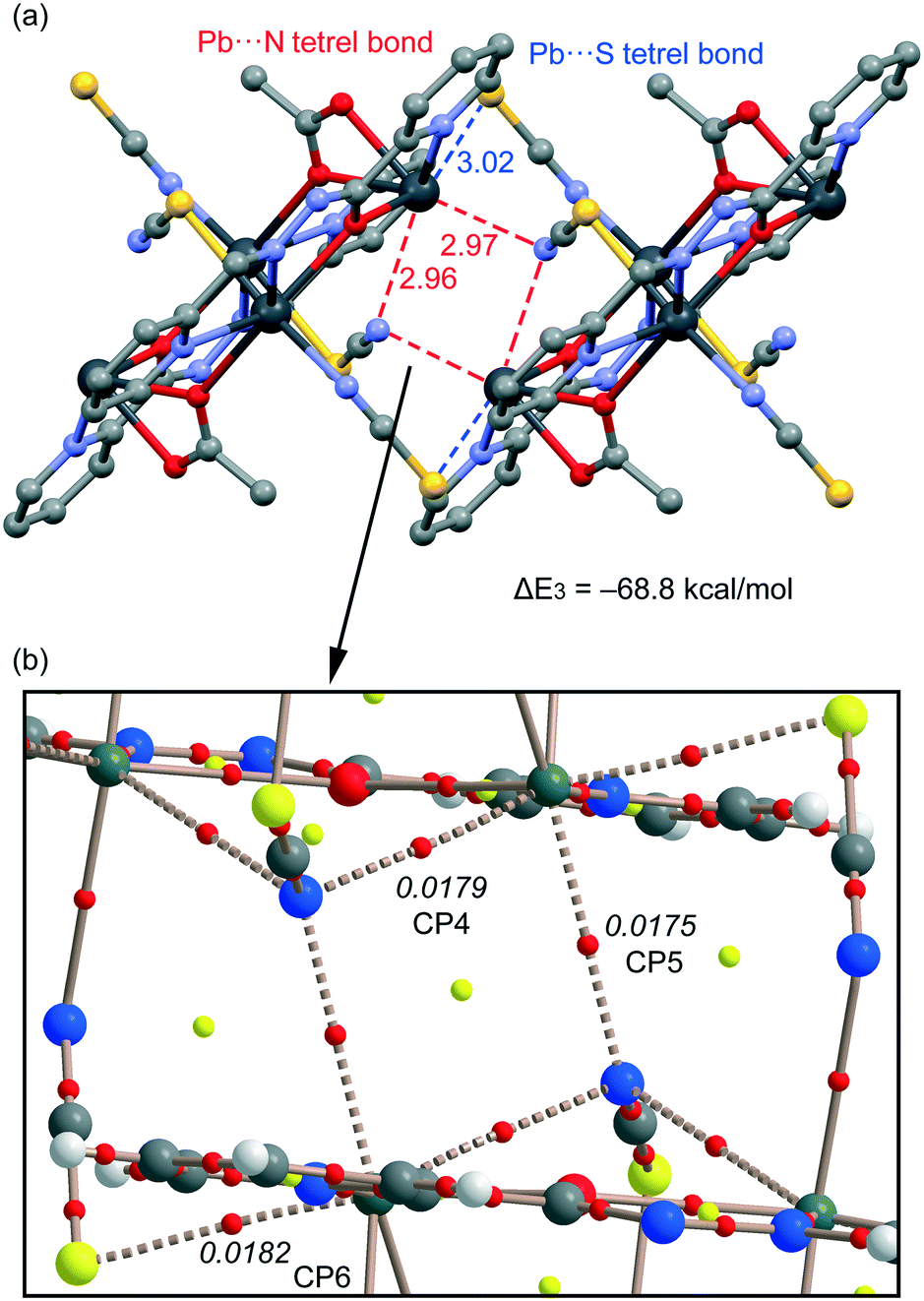
For compound 3, we have studied theoretically the interesting 1D supramolecular polymeric chain (see Fig. 4) that it is formed in the solid state. We have evaluated the interaction energy of the dimeric model extracted from the X-ray structure where six concurrent tetrel bonds are formed (see Fig. 9a). The four N⋯Pb tetrel bonds are represented in red (two of them are intramolecular; the Pb⋯N distance is 2.96 Å). In these interactions, the SCN ligand is coordinated to the Pb atom by the S atom (coordination bond) which establishes the tetrel bonding interaction (non-covalent bond) by the N end of the pseudohalide. In the S⋯Pb tetrel bonds (blue dashed lines), the pseudohalide is coordinated to the Pb atom at the N end. The interaction energy of this dimer is very large (ΔE3 = −68.8 kcal mol−1), thus confirming the importance of this interaction governing the solid state architecture of compound 3. We have also carried out the AIM analysis in this dimer and highlighted a partial view of the distribution in Fig. 9b. All the tetrel bonds are characterized by a bond CP and bond path connecting the Pb atoms to the N/S atom (ρBCP = 0.0175–0.0182 a.u.). These large values of ρ at the bond CPs are consistent with the strong binding energy of the self-assembled dimer.
 |
| | Fig. 9 (a) The dimer formed in the crystal packing of 3. (b) The AIM distribution of the bond and ring critical points (red and yellow spheres, respectively) and bond paths corresponding to the Pb⋯S/N interactions. The values of ρ(r) (in a.u.) at the bond CPs are given in italics. | |
Finally, in compound 4, we have studied theoretically the charge-assisted tetrel bonding interaction between the outer-sphere thiocyanate and the Pb(II) complex (see Fig. 5). We have first computed the MEP surface (see Fig. 10a), and a strong σ-hole is observed at the extension of the N–Pb bond (+75 kcal mol−1). This strong σ-hole is due to the total charge of +1 of this moiety. Consequently, the interaction energy computed for the tetrel bonded complex with the SCN− counterion is very large (ΔE4 = −64.9 kcal mol−1) for a single tetrel bond. We have used the AIM analysis and distribution of CPs to confirm the existence of the S⋯Pb tetrel bond (see Fig. 10b). The σ-hole interaction is characterized by the presence of a bond CP (ρ = 0.0193 a.u.) and a bond path inter-connecting the S and Pb atoms (see Fig. 10b). The value of ρ at the bond CP is very large, which is in agreement with the strong (charge-assisted) interaction energy.
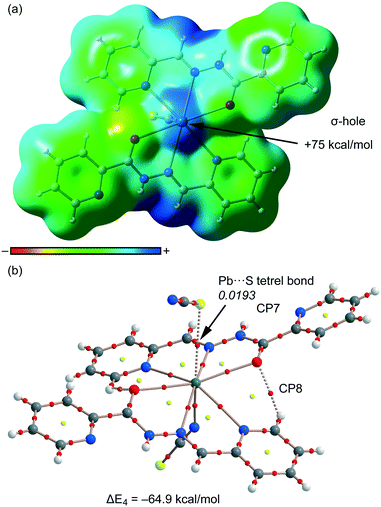 |
| | Fig. 10 (a) MEP surface (isosurface = 0.001 a.u.) of compound 4. MEP values at selected points of the surface are indicated. A theoretical model used to evaluate the binding energy. (b) AIM distribution of the bond and ring critical points (red and yellow spheres, respectively) and bond paths for the tetrel bonded complex 4. The values of ρ(r) at the bond CPs are given in italics. | |
In Table 2, we gather the charge electron density and the Laplacian and ellipticity values [ρ(r), ∇2ρ(r) and ε, respectively] at the bond CPs that characterize the non-covalent H-bonding interactions described above and labelled in Fig. 7–10. The value of ρ(r) can be related to the strength of the interaction.29 The density values at the bond CPs reveal that the Pb⋯S interaction in 4 is the strongest one which is in agreement with its large interaction energy. Moreover, the values of ρ(r) in compounds 3 and 1 that characterize the tetrel bonds are also large, which are in line with the strong tetrel bonding interactions computed in these complexes. Finally, complex 2 presents and corresponds to the weakest complex (ΔE2 = −5.8 kcal mol−1).
Table 2 AIM parameters (ρ, ∇2ρ and ε, a.u.) at the bond CPs (see Fig. 7–10 for labelling) at the M06-2X/def2.TZVP level of theory
| CP label, Compd |
102 × ρ |
102 × ∇2ρ |
102 × ε |
| CP1 (Pb⋯N), 1 |
1.88 |
4.75 |
12.1 |
| CP2 (H⋯O), 1 |
1.35 |
4.14 |
3.72 |
| CP3 (Pb⋯I), 2 |
1.34 |
2.53 |
0.99 |
| CP4 (Pb⋯N), 3 |
1.79 |
28.9 |
7.64 |
| CP5 (Pb⋯N), 3 |
1.75 |
31.9 |
6.89 |
| CP6 (Pb⋯S), 3 |
1.82 |
4.16 |
1.10 |
| CP7 (Pb⋯S), 4 |
1.93 |
4.18 |
0.25 |
| CP8 (H⋯O), 4 |
1.05 |
4.12 |
9.01 |
4. Concluding remarks
In conclusion, we report the syntheses and structural characterization of four new Pb2+ complexes with nicotinoylhydrazone and picolinoylhydrazone-based ligands. All the compounds 1–4 exhibit relevant Pb⋯X (X = N, S, I) tetrel bonding interactions in the solid state which have been described and characterized using DFT calculations. The energies associated with the interactions have also been evaluated using DFT calculations. In general, the non-covalent Pb⋯X interactions range from moderate to strong depending on the Pb2+ environment. The electrostatic effects, as shown by the MEP analysis, are very important to describe this interaction. Finally, the results included herein reveal that non-covalent tetrel bonding interactions are crucial to understand the solid-state architecture of organic–inorganic materials systems that contain hemidirectionally coordinated Pb2+ metal centers and organic aromatic molecules.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to the University of Maragheh for the financial support of this research. S. K. Seth is grateful to the SERB-DST (Govt. of India) for Overseas Postdoctoral Fellowship (SB/OS/PDF-524/2015-16). The publication was also prepared with the support of the “RUDN University Program 5-100”. AB and AF thank MINECO/AEI of Spain (projects CTQ2014-57393-C2-1-P and CTQ2017-85821-R FEDER funds) for funding and the CTI (UIB) for the free allocation of computer time.
References
-
(a) Y. Wang, L. J. Zhou, M. L. Feng and X. Y. Huang, Dalton Trans., 2012, 41, 6689 RSC; C. S. Liu, X. S. Shi, J. R. Li, J. J. Wang and X. H. Bu, Cryst. Growth Des., 2006, 6, 656 Search PubMed;
(b) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS PubMed;
(c) S. G. Telfer and R. Kuroda, Coord. Chem. Rev., 2003, 242, 33 CrossRef CAS;
(d) S. A. Barnett and N. R. Champness, Coord. Chem. Rev., 2003, 246, 145 CrossRef CAS.
- Y.-F. Hsu, H.-L. Hu, C.-J. Wu, C.-W. Yeh, D. M. Proserpio and J.-D. Chen, CrystEngComm, 2009, 11, 168 RSC.
- J. Yang, B. Wu, F. Zhuge, J. Liang, C. Jia, Y.-Y. Wang, N. Tang, X.-J. Yang and Q.-Z. Shi, Cryst. Growth Des., 2010, 10, 2331 CAS.
- F. F. B. J. Janssen, L. P. J. Veraart, J. M. M. Smits, R. de Gelder and A. E. Rowan, Cryst. Growth Des., 2011, 11, 4313 CAS.
-
(a)
G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, Oxford, 1997 RSC; Y. Xia, S. Li, B. Wu, Y. Liua and X.-J. Yanga, CrystEngComm, 2011, 13, 5763 RSC;
(b) P. Dechambenoit, S. Ferlay, M. W. Hosseini and N. Kyritsakas, Chem. Commun., 2007, 4626 RSC;
(c) W. Li, A. Thirumurugan, P. T. Barton, Z. Lin, S. Henke, H. H.-M. Yeung, M. T. Wharmby, E. G. Bithell, C. J. Howard and A. K. Cheetham, J. Am. Chem. Soc., 2014, 136, 7801 CrossRef CAS PubMed;
(d) J. M. Roberts, B. M. Fini, A. A. Sarjeant, O. K. Farha, J. T. Hupp and K. A. Scheidt, J. Am. Chem. Soc., 2012, 134, 3334 CrossRef CAS PubMed; R. S. Forgan, R. J. Marshall, M. Struckmann, A. B. Bleine, D.-L. Long, M. C. Bernini and D. Fairen-Jimenez, CrystEngComm, 2015, 17, 299 Search PubMed;
(e) M. Servati-Gargari, G. Mahmoudi, S. R. Batten, V. Stilinović, D. Butler, L. Beauvais, W. S. Kassel, W. G. Dougherty and D. VanDerveer, Cryst. Growth Des., 2015, 15, 1336 CrossRef CAS.
-
(a) P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178 RSC;
(b) A. Bauzá, T. J. Mooibroek and A. Frontera, ChemPhysChem, 2015, 16, 2496 CrossRef PubMed;
(c) A. Bauzá and A. Frontera, Angew. Chem., Int. Ed., 2015, 54, 7340 CrossRef PubMed.
-
(a) J. S. Murray, K. E. Riley, P. Politzer and T. Clark, Aust. J. Chem., 2010, 63, 1598 CrossRef CAS;
(b) P. Politzer and J. S. Murray, ChemPhysChem, 2013, 14, 278 CrossRef CAS PubMed;
(c) P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748 RSC;
(d) P. Politzer, K. E. Riley, F. A. Bulat and J. S. Murray, Comput. Theor. Chem., 2012, 998, 2 CrossRef CAS;
(e) A. Bauzá, I. Alkorta, A. Frontera and J. Elguero, J. Chem. Theory Comput., 2013, 9, 5201 CrossRef PubMed;
(f) G. Sánchez-Sanz, C. Trujillo, I. Alkorta and J. Elguero, Phys. Chem. Chem. Phys., 2014, 16, 15900–15909 RSC.
-
(a) A. Bauzá, T. J. Mooibroek and A. Frontera, Angew. Chem., Int. Ed., 2013, 52, 12317 CrossRef PubMed;
(b) S. J. Grabowski, Phys. Chem. Chem. Phys., 2014, 16, 1824 RSC;
(c) A. Bauzá, T. J. Mooibroek and A. Frontera, Chem. – Eur. J., 2014, 20, 10245 CrossRef PubMed;
(d) E. C. Escudero-Adán, A. Bauzá, A. Frontera and P. Ballester, ChemPhysChem, 2015, 16, 2530 CrossRef PubMed; A. Bauzá, T. J. Mooibroek and A. Frontera, Phys. Chem. Chem. Phys., 2014, 16, 19192 Search PubMed;
(e) A. Bauzá, T. J. Mooibroek and A. Frontera, Chem. Commun., 2014, 50, 12626 RSC;
(f) A. Bauzá, T. J. Mooibroek and A. Frontera, Chem. Rec., 2016, 16, 473 CrossRef PubMed;
(g) J. S. Murray, P. Lane and P. Politzer, J. Mol. Model., 2009, 15, 7239 Search PubMed;
(h) A. Bundhun, P. Ramasami, J. S. Murray and P. Politzer, J. Mol. Model., 2013, 19, 2739 CrossRef CAS PubMed;
(i) R. S. Ruoff, T. Emilsson, A. I. Jaman, T. C. Germann and H. S. Gutowsky, J. Chem. Phys., 1992, 96, 3441 CrossRef CAS;
(j) R.-D. Urban, G. Rouillé and M. Takami, J. Mol. Struct., 1997, 413–414, 511 CrossRef CAS;
(k) I. Alkorta, I. Rozas and J. Elguero, J. Phys. Chem. A, 2001, 105, 743 CrossRef CAS.
-
(a) M. Servati Gargari, V. Stilinović, A. Bauzá, A. Frontera, P. McArdle, D. Van Derveer, S. W. Ng and G. Mahmoudi, Chem. – Eur. J., 2015, 21, 17951–17958 CrossRef CAS PubMed;
(b) G. Mahmoudi, A. Bauza, A. Frontera, P. Garczarek, V. Stilinovic, A. M. Kirillov, A. Kennedy and C. Ruiz-Perez, CrystEngComm, 2016, 18, 5375–5385 RSC;
(c) G. Mahmoudi, A. Bauza, M. Amini, E. Molins, J. T. Mague and A. Frontera, Dalton Trans., 2016, 45, 10708–10716 RSC;
(d) G. Mahmoudi, V. Stilinovic, A. Bauza, A. Frontera, A. Bartyzel, C. Ruiz-Perez and A. M. Kirillov, RSC Adv., 2016, 6, 60385–60393 RSC;
(e) G. Mahmoudi, L. Dey, H. Chowdhury, A. Bauza, B. K. Ghosh, A. M. Kirillov, S. K. Seth, A. V. Gurbanov and A. Frontera, Inorg. Chim. Acta, 2017, 461, 192–205 CrossRef CAS;
(f) S. Roy, M. G. B. Drew, A. Bauzá, A. Frontera and S. Chattopadhyay, New J. Chem., 2018, 42, 6062–6076 RSC.
- G. Mahmoudi, A. Bauzá and A. Frontera, Dalton Trans., 2016, 45, 4965–4969 RSC.
- A. A. Khandar, B. K. Ghosh, C. Lampropoulos, M. S. Gargari, V. T. Yilmaz, K. Bhar, S. A. Hosseini-Yazdi, J. M. Cain and G. Mahmoudi, Polyhedron, 2015, 85, 467 CrossRef CAS.
-
SAINT Plus, Data Reduction and Correction Program, v. 6.01, Bruker AXS, Madison, Wisconsin, USA, 1998 Search PubMed.
-
SADABS v. 2.01, Bruker/Siemens Area Detector Absorption Correction Program, Bruker AXS, Madison, Wisconsin, USA, 1998 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- W. T. Pennington, J. Appl. Crystallogr., 1999, 32, 1028 CrossRef CAS.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378 RSC.
-
(a) J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814 RSC;
(b) M. A. Spackman, J. J. McKinnon and D. Jayatilaka, CrystEngComm, 2008, 10, 377 CAS;
(c) M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19 RSC;
(d) F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129 CrossRef CAS.
-
(a) S. K. Seth, I. Saha, C. Estarellas, A. Frontera, T. Kar and S. Mukhopadhyay, Cryst. Growth Des., 2011, 11, 3250 CrossRef CAS;
(b) P. Manna, S. K. Seth, A. Das, J. Hemming, R. Prendergast, M. Helliwell, S. R. Choudhury, A. Frontera and S. Mukhopadhyay, Inorg. Chem., 2012, 51, 3557 CrossRef CAS PubMed;
(c) S. K. Seth, CrystEngComm, 2013, 15, 1772 RSC;
(d) M. Mitra, S. K. Seth, S. R. Choudhury, P. Manna, A. Das, M. Helliwell, A. Bauzá, A. Frontera and S. Mukhopadhyay, Eur. J. Inorg. Chem., 2013, 4679 CrossRef CAS;
(e) P. Manna, S. R. Choudhury, M. Mitra, S. K. Seth, M. Helliwell, A. Bauzá, A. Frontera and S. Mukhopadhyay, J. Solid State Chem., 2014, 220, 149 CrossRef CAS;
(f) S. K. Seth, A. Bauzá and A. Frontera, CrystEngComm, 2018, 20, 746 RSC.
-
(a) M. A. Spackman and P. G. Byrom, Chem. Phys. Lett., 1997, 267, 215 CrossRef CAS;
(b) J. J. McKinnon, A. S. Mitchell and M. A. Spackman, Chem. – Eur. J., 1998, 4, 2136 CrossRef CAS.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627 CrossRef PubMed.
-
(a) A. L. Rohl, M. Moret, W. Kaminsky, K. Claborn, J. J. Mckinnon and B. Kahr, Cryst. Growth Des., 2008, 8, 4517 CrossRef CAS;
(b) A. Parkin, G. Barr, W. Dong, C. J. Gilmore, D. Jayatilaka, J. J. Mckinnon, M. A. Spackman and C. C. Wilson, CrystEngComm, 2007, 9, 648 RSC;
(c) S. K. Seth, D. Sarkar, A. D. Jana and T. Kar, Cryst. Growth Des., 2011, 11, 4837 CrossRef CAS;
(d) S. K. Seth, D. Sarkar and T. Kar, CrystEngComm, 2011, 13, 4528 RSC;
(e) M. Mitra, P. Manna, S. K. Seth, A. Das, J. Meredith, M. Helliwell, A. Bauzá, S. R. Choudhury, A. Frontera and S. Mukhopadhyay, CrystEngComm, 2013, 15, 686 RSC;
(f) S. K. Seth, J. Mol. Struct., 2014, 1070, 65 CrossRef CAS;
(g) M. Mitra, P. Manna, A. Bauzá, P. Ballester, S. K. Seth, S. R. Choudhury, A. Frontera and S. Mukhopadhyay, J. Phys. Chem. B, 2014, 118, 14713 CrossRef CAS PubMed.
-
S. K. Wolff, D. J. Grimwood, J. J. McKinnon, D. Jayatilaka and M. A. Spackman, CrystalExplorer 3.1, The University of Western Australia, Perth, Australia, 2012 Search PubMed.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
-
T. A. Keith, AIMAll (Version 13.05.06), TK Gristmill Software, Overland Park KS, USA, 2013 Search PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
-
(a) I. Mata, I. Alkorta, E. Molins and E. Espinosa, Chem. – Eur. J., 2010, 16, 2442–2452 CrossRef CAS PubMed;
(b) E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS;
(c) E. Espinosa, C. Lecomte and E. Molins, Chem. Phys. Lett., 1999, 300, 745–748 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1588614–1588617 contain the supplementary crystallographic data for complexes 1–4. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ce00110c |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Saikat Kumar
Seth
*a,
Saikat Kumar
Seth