
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymorphism of chlorpropamide on liquid-assisted mechanical treatment: choice of liquid and type of mechanical treatment matter†
Nadia
Bouvart‡
 ab,
Roland-Marie
Palix‡
ab,
Sergey G.
Arkhipov
ac,
Ivan A.
Tumanov
ac,
Adam A. L.
Michalchuk
*ade and
Elena V.
Boldyreva
*ac
ab,
Roland-Marie
Palix‡
ab,
Sergey G.
Arkhipov
ac,
Ivan A.
Tumanov
ac,
Adam A. L.
Michalchuk
*ade and
Elena V.
Boldyreva
*ac
aNovosibirsk State University, Novosibirsk, Russian Federation. E-mail: i.tumanov.ssc@mail.ru; eboldyreva@yahoo.com
bESCOM, 1 allée du réseau J.-M. Buckmaster, 60200 Compiègne, France. E-mail: nadia.bouvart@live.fr
cInstitute of Solid State Chemistry and Mechanochemistry, SB RAS, Novosibirsk, Russian Federation
dEaStChem School of Chemistry, University of Edinburgh, Edinburgh, UK. E-mail: adam.michalchuk@ed.ac.uk
eEPSRC Centre for Continuous Manufacturing and Crystallisation (CMAC), UK
First published on 12th February 2018
Abstract
Different types of mechanical treatment (tableting, grinding, milling, etc.) are important technological operations in the pharmaceutical industry. Solid materials are not merely comminuted during such treatment, but can undergo polymorphic transitions and amorphisation. A liquid is often added to the solid sample on purpose, e.g. in order to facilitate comminution. In many cases, liquid is present inadvertently, either as a result of trace solvent following up-stream operations, or sorbed from the atmosphere. This work explores the liquid-assisted mechanical treatment of two metastable forms of a model pharmaceutical compound chlorpropamide (CPA), namely the β- and ε-forms. We investigate the stability of these polymorphs to mechanical treatment in the presence of a series of fluids that have affinity for different fragments of the CPA molecule. In addition to variation in fluids, the effect of different mechanical treatment is explored, employing model shear and restricted impact devices, alongside grinding in a mortar. CPA offers a clear example of the drastically different results of shear and impact on mechanochemical polymorphism, on the importance of the careful selection of solvent for liquid-assisted treatment, and on the possible role of even trace quantities of solvent. Further, this work demonstrates the importance of the starting polymorph in determining the outcome of a mechanochemical process. All of these factors can have notable consequence on the outcome of technological operations that include mechanical treatment.
Introduction
Pharmaceutical compounds are often administered in the form of a solid powder. The crystalline structure of these powder particles plays an important role in determining the physico-chemical properties of the pharmaceutical material, particularly the dissolution dynamics.1–3 Different crystalline forms, polymorphs, can display vastly different properties.4 The ability to control the crystalline form is therefore critical to ensure the integrity of materials. The interrelation between crystal structure and physico-chemical properties has led to considerable work in controlling crystal form through selective crystallisation of polymorphs, or through the design of multi-component materials.1–9 This has typically been done through solution-based processes, although mechanochemical approaches, particularly milling, can be used.9–11 For pharmaceutical applications, one generally aims to produce a metastable phase of the crystalline material, which displays higher solubility than the thermodynamically stable form.A variety of milling processes are employed in the industrial processing of pharmaceutical materials.9–18 At present this is generally aimed at reducing particle size with enhanced solubility properties. While dry milling techniques are possible, the resulting powders are often highly metastable and the powder particles agglomerate quickly. Instead, wet milling techniques have been shown to be more promising.10,19–30 In these cases, milling is performed on a suspension of particles in either aqueous or non-aqueous solvents. The selection of solvent is often done so as to ensure minimal dissolution of the system being milled, ease of drying, and to ensure any residual solvent is not toxic. The wet-milled mixtures are investigated for possible contamination by corroded material from the milling equipment, and for residual solvent. However, little concern is given towards the effects of the liquid on the integrity of the crystalline phase itself. This is particularly important, as the solid materials are not merely comminuted during such treatment, but can change or completely lose their crystal structure (polymorphic transitions and amorphisation, respectively), even if no chemical changes of the molecular structure occurs. This phenomenon can be termed as “structural instability”. Even if a liquid is not added on purpose, it is often present in trace quantities following previous operations, and can therefore inadvertently induce structural changes.
In the present work we explore the structural stability of a model pharmaceutical material, chlorpropamide (CPA), a sulfonylurea compound used for treatment of type 2 diabetes mellitus31 (Fig. 1).
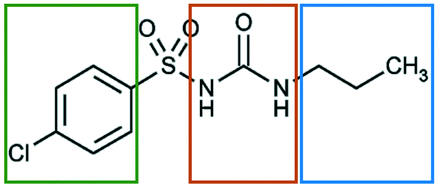 | ||
| Fig. 1 Structural formula of a CPA molecule. The fragments differing in affinity to different solvents are shown by frames of different colors. | ||
CPA is well known to be extremely prone to polymorphism.32–41 Although the α-form is thermodynamically stable under ambient conditions, other forms can be obtained, often concomitantly, as dominant phases or as impurities.35–39 These metastable phases persist in mixtures under ambient conditions, recrystallising only slowly to the stable form, hastened by humid atmosphere. Obtaining any metastable phase as a pure phase remains immensely challenging. Only a method for reproducibly obtaining ε-CPA is known.39 The β- and ε-forms of CPA are the most interesting for pharmaceutical applications, displaying the highest dissolution rates across the series of isolable CPA forms.42 A method to obtain bulk quantities of β-CPA has not been documented prior to this work.
Transitions between polymorphs of the CPA on hydrostatic compression have been shown to depend strongly on the presence of a pressure-transmitting fluid43 and on its choice.44–46 CPA thus provides an intriguing system for further analysis of the effects of different liquids on its polymorphism, including polymorphic transitions induced by mechanical treatment.
The molecular structure of CPA contains three chemically unique segments, an aliphatic tail, a carbamide group, and a halogenated aromatic (Fig. 1). Solvents have therefore been explicitly selected to target interactions with specific components of the CPA molecule. Toluene was selected to interact with the aromatic fragment, and heptane was chosen to interact with the hydrocarbon tail. Shorter hydrocarbons were not selected due to their volatilities. The hydrogen bonding solvent, ethanol, was selected to interact with the carbamide group, with similar types of interactions expected by atmospheric water. Chloroform was supposed to be able to interact more or less equally with the alkyl tail and the chlorinated aromatic ring, but not with the sulphonamide fragment or the urea core.
An earlier study on the mechanochemistry of the α- and ε-polymorphs of CPA showed no transformation when dry milled under ambient temperature in a vibratory ball mill.47 It has been documented for some other organic molecular crystals that different types of treatment (such as shear or impact) may have different or even opposing effects on a transformation.48,49 It is therefore important to compare the outcome of the transformations on different types of mechanical treatment. In the present work we used custom-built model devices48–52 to compare the effects of shear and restricted impact, and compared them with the outcome of grinding with a mortar and pestle.
The discovery of a new method to obtain pure β-CPA, reported here, enabled us to explore the stability of both β- and ε-CPA to mechanical manipulation for solid form processing, depending on the choice of added liquid and the type of mechanical treatment.
Experimental
Materials
All polymorphs were obtained starting from commercially available chlorpropamide (Sigma Aldrich, 97%). Experiments employed solvents heptane (Reachim, Russia), chloroform (N°1 Chemical Reagents, Russia), ethanol (KPF, Russia) and toluene (Reactiv, Russia). All materials were used without further purification.Growth of ε-chlorpropamide
Metastable ε-CPA was produced as described in an earlier study.39 In this study, the metastable phase was obtained by heating the commercially available α-form to 120 °C, holding for 1 h, and cooling at approximately 0.5 °C min−1 to room temperature.Growth of β-chlorpropamide
The metastable β-CPA phase has proved challenging to reliably reproduce. In the present work, a robust method for formation of pure β-CPA was developed. Following the idea of drop-wise crystallisation procedures published previously for other compounds,53 a solution of CPA in chloroform was placed drop-wise onto a pre-heated glass slide at 61 °C (hot plate WiseStir MSH-20D was used). We note that the temperature was very important to ensure that pure β-CPA was produced (ESI†). Each drop was carefully separated from neighbouring drops, ensuring that crystallites did not re-dissolve in newly added drops. Drops were left to evaporate at this temperature, resulting in formation of β-CPA crystals. Purity was confirmed by X-ray powder diffraction.Manual grinding
Manual grinding was performed with an onyx mortar and pestle at room temperature (ca. 25 °C) and relative humidity (RH) of ca. 50–60%. For each experiment, a sample of ca. 30 mg was used. A series of experiments was also performed in a glove box under a dry N2 atmosphere, with RH <10%. For LAG experiments by manual grinding, ca. 30 mg of material was ground with a single drop of liquid (ca. 20 μL), giving a liquid-to-solid ratio of η ≈ 0.67 μL mg−1.Restricted impact
Mechanochemical experiments with restricted impact treatment were performed in a custom model device, which has been used and described in previous studies.50 Samples (ca. 30 mg) were subjected to a series of periodic mechanical impacts. The kinetic energy of individual impacts was approximately 10 mJ per impact, with impact frequency of 3.33 Hz (200 per minute). Each experiment was performed under the same ambient conditions, with the same settings, for varying lengths of time. Following mechanical treatment, the entire powder sample was transferred immediately to the diffractometer for analysis by X-ray powder diffraction (XRPD). For LAG experiments by restricted impact, ca. 30 mg of material was ground with a single drop of liquid (ca. 20 μL), giving a liquid-to-solid ratio of η ≈ 0.67 μL mg−1.Shear
Shear treatment was performed in a second custom device, described previously.48 Samples (ca. 30 mg) were subjected to continuous mechanical shearing between two circular, roughened glass plates, while under the pressure of several metal weights. The estimated average pressure on the sample was 4 kPa. Mechanochemical experiments were performed under the same ambient conditions, for varying lengths of time. After each experiment the entire powder sample was transferred immediately to the diffractometer for analysis by X-ray powder diffraction (XRPD). For LAG experiments by shear, ca. 30 mg of material was ground with a single drop of liquid (ca. 20 μL), giving a liquid-to-solid ratio of η ≈ 0.67 μL mg−1.Characterisation
All solid samples were characterised by X-ray powder diffraction (XRPD) using a STOE-MP diffractometer. The diffractometer was equipped with a Cu anode (Kα1 1.5406 Å) and a bent Ge(111) monochromator. XRPD patterns were collected in transmission mode in steps of 0.015 deg, with collection time 2 s per step.Results and discussion
Atmospheric stability of metastable chlorpropamide
As initial assessment of the effects of liquids on the metastable β- and ε-phases of CPA, samples of each material were stored under low (ca. 10%) and high (ca. 90%) relative humidity (RH) atmosphere. For both phases, no obvious transformation was detected when the sample was stored at 10% RH for nine days. However, for samples stored under ca. 90% RH polymorphic conversions were observed. In the case of ε-CPA, the sample was found to have undergone notable conversion to the thermodynamically stable α-phase within four days. In contrast, the sample of β-CPA displayed similar conversion to the α-phase only after seven days at ca. 90% RH (ESI†).This strongly suggests that the transformation of the metastable polymorphs to the stable form proceeds by recrystallisation in atmospheric moisture. Interestingly, while the molecular conformation in α-CPA more closely resembles that of β-CPA35 (Fig. 2), the molecular packing in the crystal structure of ε-CPA37 is more similar to that in α-CPA34 (Fig. 3). It is therefore logical to find that the latter transformation should occur more rapidly, in the presence of only small quantities of solvent.
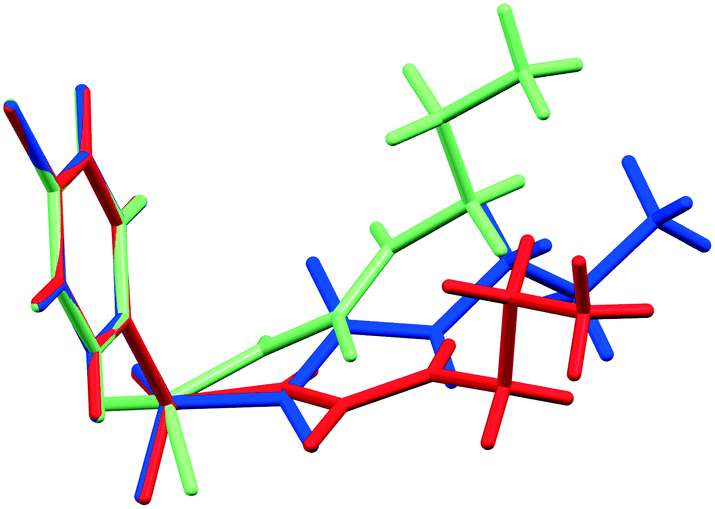 | ||
| Fig. 2 Overlay of the asymmetric CPA molecule in the α-CPA (blue), β-CPA (red), and ε-CPA (green). | ||
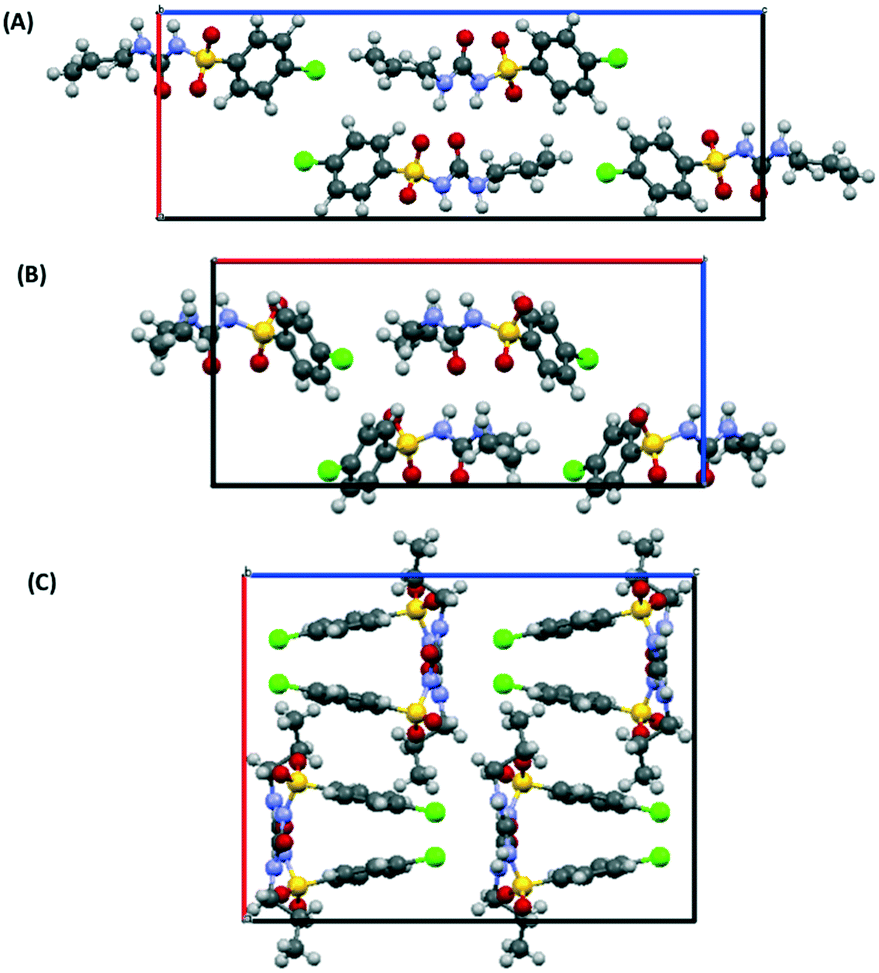 | ||
| Fig. 3 Molecular packing in the crystal structures of the (A) α-, (B) ε- and (C) β-forms of CPA. Structures are viewed along the crystallographic c-axis. Structural data from ref. 34, 35 and 37. | ||
Previous work on the CPA polymorphs has shown that both α- and β-polymorphs transform into the ε-form on slow heating prior to melting, with the solid-state transformation of the β-form into the ε-form being kinetically hindered. The β-polymorph can therefore melt without conversion to the ε-form if heated quickly.38,39 At ambient conditions the stability of the forms decreases as α > ε > β (ref. 39) under ambient pressure. Recrystallisation of CPA in atmospheric moisture therefore follows according to Ostwald's rule of stages.54–56
Mechanical treatment of ε- and β-chlorpropamide under dry or humid conditions
Initial manual grinding of the pure phases of the β- and ε-forms of CPA under ambient conditions showed slow conversion to the stable α-phase in each case (ESI†). This was to be expected given the response of each material to humid summer atmosphere, and highlights the influence of humidity on mechanochemical reactions as well as the accelerating effect of grinding on solid state processes. In contrast, conducting these same grinding processes under a dry atmosphere in a glove box or in winter47 (<20% RH) resulted in no, or only traces of transformation (Fig. 4 and ESI†). This is an example of the seasonal irreproducibility of mechanochemical transformations that are sensitive to the atmospheric humidity, termed inadvertent liquid-assisted grinding.57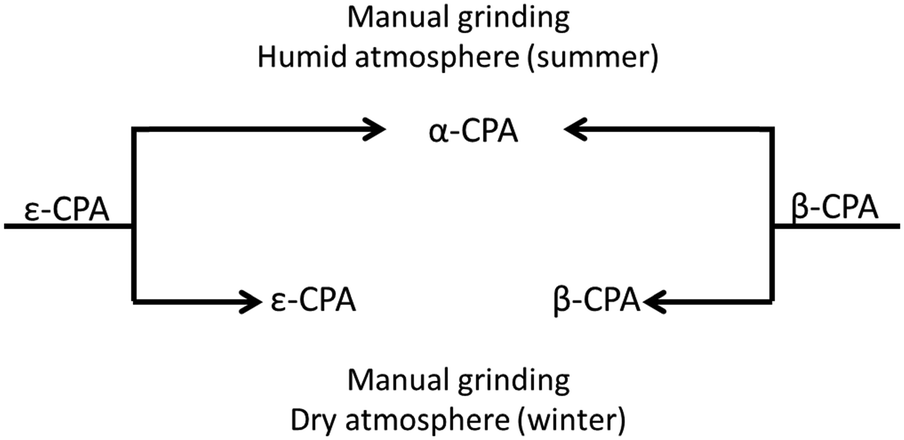 | ||
| Fig. 4 Schematic influence of humidity on CPA phase conversion. Both forms convert to α-CPA when manually ground under a humid atmosphere. No conversion is found under dry atmosphere. | ||
Liquid-assisted grinding (LAG) of ε- and β-chlorpropamide
A summary of the transformations of both metastable polymorphs of CPA is given in Fig. 5.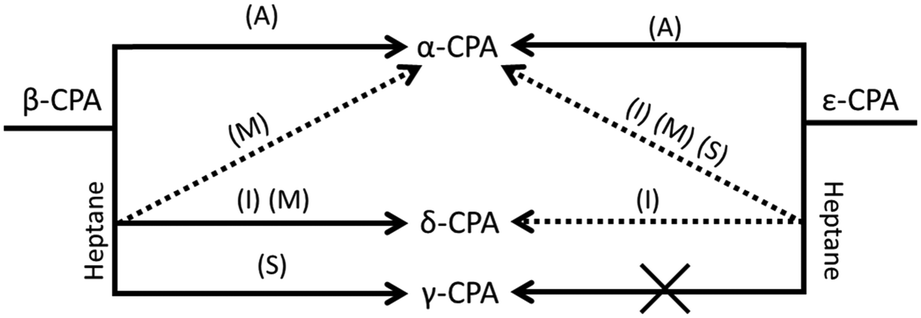 | ||
| Fig. 5 Schematic summary of the mechanically-induced transformations of ε- and β-CPA. (A) Transformation observed under all types of mechanical treatment in the presence of moisture, ethanol, chloroform or toluene. Specific reactivity under restricted impact (I), manual grinding (M) or shear (S) are indicated for LAG with heptane. | ||
Liquid-assisted grinding (LAG) of ε-chlorpropamide
As with manual ‘dry’ grinding under atmospheric humidity, the grinding of ε-CPA in the presence of ethanol, toluene, chloroform or heptane led to gradual conversion of ε → α-CPA (ESI†). In all cases, only minimal grinding was required to observe initial signs of transformation. Industrially, this is a promising result, where any seeds of ε-CPA that may be present in the initial system (readily formed by heating), can be easily removed by wet milling.The same transformation was observed when ε-CPA was subjected to impact treatment in most cases. When impact was performed in the presence of chloroform or ethanol, complete transformation to the α-phase was observed within 30 minutes. However, residual quantities of the starting ε-phase remained after 30 minutes when impact was performed in the presence of toluene (ESI†).
Interestingly, the addition of heptane to the impact reaction occasionally induced formation of small quantities of an impurity phase. In such small quantities it is difficult to assign this phase unambiguously (ESI†). However, it appears that the impurity phase can be either the α- or closely related δ-phase, depending on the experiment. Despite the minor impurity forming, the majority of powder remains as the original ε-form, Fig. 6. The possibility to control selective formation of the α- or δ-forms under restricted impact treatment with heptane will be the subject of further investigation, but may be associated with residual atmospheric moisture in some cases. We note that in both the ε and α-phases, the alkyl tail of the CPA molecule is linear, but bent in the δ-form. The formation of δ-CPA in the presence of heptane corresponds to the known crystallisation of the δ-form from heptane–ethyl acetate solution,37 believed to be associated with interaction of the solvent alkyl chains with the CPA tail on recrystallisation.45 This suggests that wet mechanochemical processing proceeds via solution-phase intermediates.
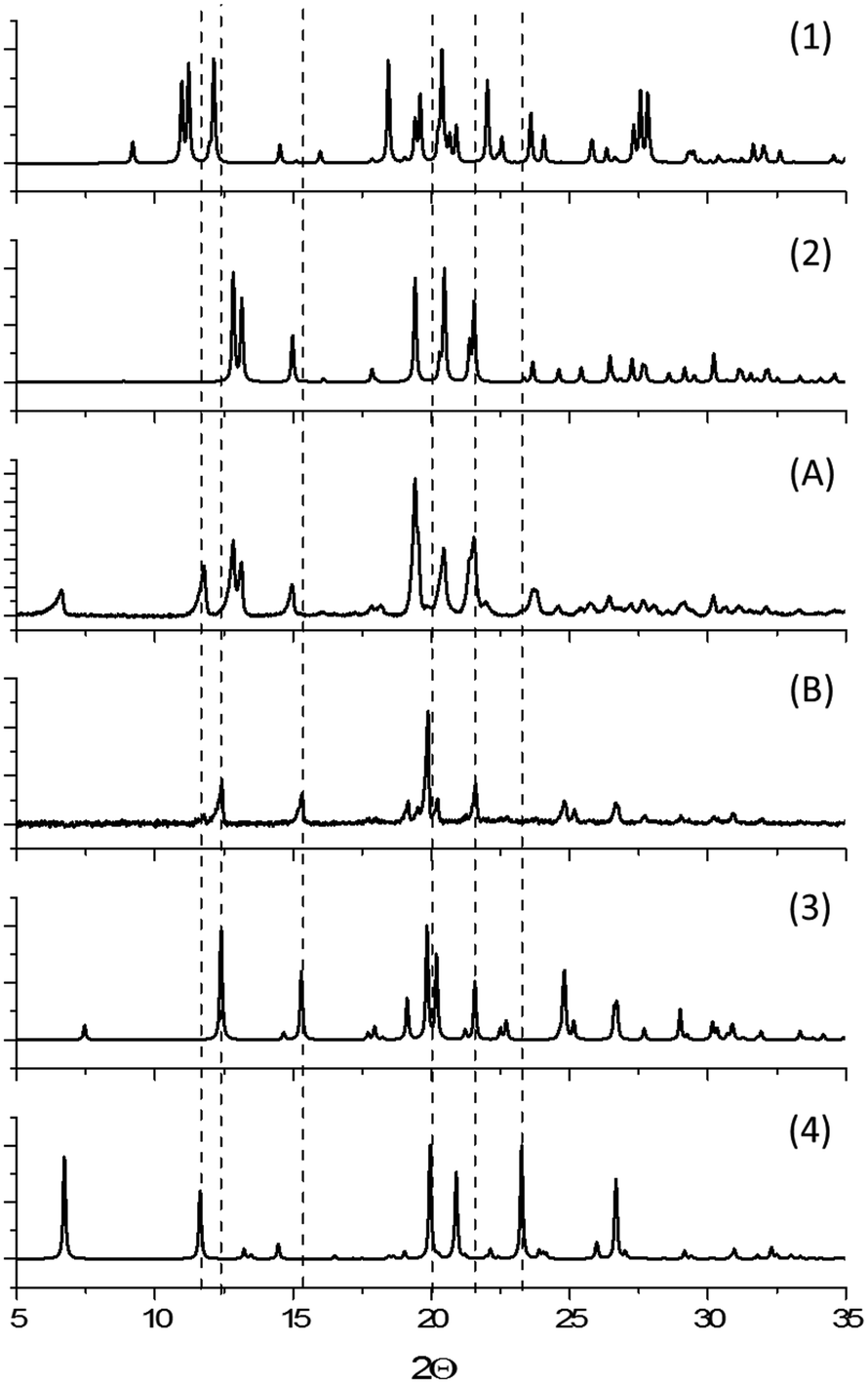 | ||
| Fig. 6 (A) XRPD pattern of ε-CPA after impact treatment with heptane, (B) XRPD pattern of β-CPA converted into γ-form after shear treatment with heptane, and simulated XRPD patterns for (1) β-CPA, (2) ε-CPA, (3) γ-CPA and (4) δ-CPA. | ||
Pure shear treatment under atmospheric conditions led to conversion of the ε-form towards the thermodynamic α-phase. This same transformation was also observed in the presence of all tested solvents, including heptane (ESI†), and is consistent with the results of manual grinding. However, this result differs from that obtained by restricted impact, where only traces of conversion were observed when conducted in the presence of heptane. This clearly demonstrates the notable differences in the effects of impact and shear mechanical treatment on the transformation of solids.48
Liquid-assisted grinding (LAG) of β-chlorpropamide
The effect of mechanical treatment on β-CPA was different from that observed for the ε-phase. Manual grinding of the β-form in the presence of toluene, ethanol or chloroform led to conversion of β-CPA to the α-polymorph in less than 30 minutes. Again, this suggests that any polymorph impurities of CPA can be readily removed by wet milling in these solvents, provided the α-phase is targeted. Instead, manual grinding in the presence of heptane led to a mixture of CPA polymorphs. Clear evidence of the α- and δ-forms is evident within only 15 minutes of grinding (ESI†). Rietveld refinement suggesting a nearly 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 mixture of the two phases is formed at this stage, with α-CPA being dominant. Only traces of β-CPA remain in this mixture. Again, this is consistent with the ability of the δ-form to be produced from heptane–ethyl acetate solutions.37
:30 mixture of the two phases is formed at this stage, with α-CPA being dominant. Only traces of β-CPA remain in this mixture. Again, this is consistent with the ability of the δ-form to be produced from heptane–ethyl acetate solutions.37
The impact treatment of β-CPA in the presence of ethanol, toluene and chloroform led to rapid conversion to the α-form (ESI†). However, in contrast to impact treatment of ε-CPA with heptane, treatment of β-CPA under these conditions led to near complete conversion to the δ-phase in 20 minutes of treatment, Fig. 6. The purity of the δ-phase continued to grow with increased treatment time (ESI†).
Shear treatment of β-CPA under atmospheric conditions, or in the presence of ethanol, toluene and chloroform led to conversion towards the α-form. Instead, shear treatment of β-CPA in the presence of heptane led to unmistakable conversion of the material into the γ-form, Fig. 6. This is in striking contrast to the effects of impact treatment on the β-form in the presence of the same solvent, and is another example (in addition to those reported before48,49) of drastically different effects of shear and impact on mechanically induced processes.
Both metastable polymorphs are therefore found to react differently under the same mechanical treatment. This offers a new dimensionality to the design and control of mechanochemical reactions, where not only must one control the solvent used for LAG, but also the starting polymorphic form. Further, as found in the case of β-CPA, it is not sufficient to control only the solvent and the polymorphic form. One must also give thought to the type of mechanical treatment to be used, when designing a mechanochemical process.
Conclusions
Mechanochemical methods are crucial for the processing of powder materials. In industry, these processes are generally used for the reduction of particle size, with wet milling being most commonly used. The presence of solvent can have drastic influence on the outcome of the mechanical treatment, and can greatly affect the nature of the resulting material. This has been identified for a number of systems, and is of growing importance.58–65 With CPA, mechanically influenced transformations are found to be solution-mediated, and are inhibited for both the β- and ε-forms when conducted in dry atmosphere. In all cases, the use of water (atmospheric), toluene, chloroform or ethanol leads to conversion of CPA towards the thermodynamically stable α-form. However, conducting mechanical treatment in the presence of heptane, expected to interact with the alkyl tail of CPA molecules, has considerably different effects.Restricted impact of ε-CPA in the presence of heptane led to nearly no conversion of the material. Instead, shear treatment in this solvent was accompanied by complete conversion to the α-form. The effects of heptane on the mechanochemistry of β-CPA were notably different. Restricted impact of β-CPA in the presence of heptane led to near complete conversion to the δ-form. However, shear treatment of β-CPA with heptane resulted in conversion to the γ-phase. Interestingly, on hydrostatic compression in a similar alkane fluid (pentane–isopentane mixture), β-CPA transforms into pure γ-CPA if no other phases are present in the system. However, if seeds of δ-CPA are present, these seeds grow in size (although no new δ-CPA crystals nucleate), concomitantly with the fast nucleation and growth of new γ-CPA crystals.45 This complex interplay of kinetics and thermodynamics is likely responsible for the drastically different results observed for the mechanochemistry of β-CPA.
Dissolution and re-crystallisation play a critical role in the mechanically-induced polymorphism of CPA. The same is true for many other organic systems. Many factors can affect the outcome of this recrystallisation, and thus the resulting product of the reaction. The present study highlights the importance of the starting polymorph, the choice of fluid, and the type of mechanical treatment (shear, impact, or hydrostatic compression) in designing a mechanochemical experiment. The design of mechanochemical processes clearly requires careful selection of all of these factors.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Financial support: Russian Foundation of Basic Research (RFBR), grant 16-33-60086 for I. T., Russian Ministry of Science and Education (S. G. and A. M.), an Edinburgh Global Research Scholarship (A. M.), EPSRC CMAC EP/I033459/1 (A. M.), project 0301-2018-0007 of the ISSCM SB RAS (E. V. B.). R. M. P. and N. B. participated in this work during their summer training course at Novosibirsk State University. They thank ESCOM for providing this opportunity to undertake an internship in Novosibirsk and are grateful to the Région Picardie of France whose funding made it possible. A. M. thanks Prof C. R. Pulham for on-going support. S. A. thanks T. N. Drebushchak for helpful advises. Additional thanks are given to Prof. V. V. Boldyrev for fruitful discussions. Final thanks are given to the reviewers for many helpful comments.Notes and references
- M. Pudipeddi and A. Serajuddin, J. Pharm. Sci., 2005, 94(5), 929–939 CrossRef CAS PubMed.
- N. Chieng, T. Rades and J. Aaltonen, J. Pharm. Biomed. Anal., 2011, 55(4), 618–644 CrossRef CAS PubMed.
- A. Avdeef, E. Fuguet, A. Llinàs, C. Ràfols, E. Bosch, G. Völgyi, T. Verbić, E. Boldyreva and K. Takács-Novák, Adv. Mater., 2016, 4(2), 117–178 Search PubMed.
- J. Bernstein, Cryst. Growth Des., 2011, 11(3), 632–650 CAS.
- D. J. Good and N. Rodríguez-Hornedo, Cryst. Growth Des., 2009, 9(5), 2252–2264 CAS.
- R. Thakuria, A. Delori, W. Jones, M. P. Lipert, L. Roy and N. Rodríguez-Hornedo, Int. J. Pharm., 2013, 453(1), 101–125 CrossRef CAS PubMed.
- N. Qiao, M. Li, W. Schlindwein, N. Malek, A. Davies and G. Trappitt, Int. J. Pharm., 2011, 419(1), 1–11 CrossRef CAS PubMed.
- C. C. Sun, Expert Opin. Drug Delivery, 2013, 10(2), 201–213 CrossRef CAS PubMed.
- D. Braga, L. Maini and F. Grepioni, Chem. Soc. Rev., 2013, 42(18), 7638–7648 RSC.
- E. Boldyreva, Curr. Pharm. Des., 2016, 22(32), 4981–5000 CrossRef CAS PubMed.
- A. Delori, T. Friščić and W. Jones, CrystEngComm, 2012, 14(7), 2350–2362 RSC.
- E. L. Parrott, J. Pharm. Sci., 1974, 63(6), 813–829 CrossRef CAS PubMed.
- V. V. Boldyrev, J. Mater. Sci., 2004, 39(16), 5117–5120 CrossRef CAS.
- V. V. Boldyrev, Russ. Chem. Rev., 2006, 75(3), 177 CrossRef CAS.
- I. Colombo, G. Grassi and M. Grassi, J. Pharm. Sci., 2009, 98(11), 3961–3986 CrossRef CAS PubMed.
- P. Baláž, M. Baláž and Z. Bujňáková, Chem. Eng. Technol., 2014, 37(5), 747–756 CrossRef.
- Z. H. Loh, A. K. Samanta and P. W. S. Heng, Asian J. Pharm. Sci., 2015, 10(4), 255–274 CrossRef.
- D. E. Crawford and J. Casaban, Adv. Mater., 2016, 28(27), 5747–5754 CrossRef CAS PubMed.
- E. Merisko-Liversidge, P. Sarpotdar, J. Bruno, S. Hajj, L. Wei, N. Peltier and J. Jones, Pharm. Res., 1996, 13(2), 272–278 CrossRef CAS.
- E. Merisko-Liversidge, G. G. Liversidge and E. R. Cooper, Eur. J. Pharm. Sci., 2003, 18(2), 113–120 CrossRef CAS PubMed.
- J. Hu, K. P. Johnston and R. O. Williams III, Drug Dev. Ind. Pharm., 2004, 30(3), 233–245 CrossRef CAS PubMed.
- P. Sharma, W. A. Denny and S. Garg, Int. J. Pharm., 2009, 380(1), 40–48 CrossRef CAS PubMed.
- Y. Tanaka, M. Inkyo, R. Yumoto, J. Nagai, M. Takano and S. Nagata, Chem. Pharm. Bull., 2009, 57(10), 1050–1057 CrossRef CAS PubMed.
- E. Merisko-Liversidge and G. G. Liversidge, Adv. Drug Delivery Rev., 2011, 63(6), 427–440 CrossRef CAS PubMed.
- P. Liu, X. Rong, J. Laru, B. van Veen, J. Kiesvaara, J. Hirvonen and L. Peltonen, Int. J. Pharm., 2011, 411(1), 215–222 CrossRef CAS PubMed.
- H. S. Ali, P. York, A. M. Ali and N. Blagden, J. Controlled Release, 2011, 149(2), 175–181 CrossRef CAS PubMed.
- A. Bhakay, M. Merwade, E. Bilgili and R. N. Dave, Drug Dev. Ind. Pharm., 2011, 37(8), 963–976 CrossRef CAS PubMed.
- Drug Delivery Strategies for Poorly Water-Soluble Drugs, ed. S. Scheler, D. Douroumis and A. Fahr, West Sussex, 2012, pp. 509–550 Search PubMed.
- T. Niwa and K. Danjo, Eur. J. Pharm. Sci., 2013, 50(3), 272–281 CrossRef CAS PubMed.
- A. Harter, L. Schenck, I. Lee and A. Cote, Org. Process Res. Dev., 2013, 17(10), 1335–1344 CrossRef CAS.
- A. E. Meinders, J. L. Touber and L. A. De Vries, Lancet, 1967, 290(7515), 544–546 CrossRef.
- A. P. Ayala, M. W. C. Caetano, S. B. Honorato, J. Mendes Filho, H. W. Siesler, S. N. Faudone and J. Ellena, J. Raman Spectrosc., 2012, 43(2), 263–272 CrossRef CAS.
- A. J. Cruz-Cabeza and J. Bernstein, Chem. Rev., 2014, 114(4), 2170–2191 CrossRef CAS PubMed.
- C. H. Koo, S. I. Cho and Y. H. Yeon, Arch. Pharmacal Res., 1980, 3(1), 37–49 CrossRef CAS.
- T. N. Drebushchak, N. V. Chukanov and E. V. Boldyreva, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62(10), o4393–o4395 CAS.
- T. N. Drebushchak, N. V. Chukanov and E. V. Boldyreva, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2007, 63(6), o355–o357 CAS.
- T. N. Drebushchak, N. V. Chukanov and E. V. Boldyreva, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2008, 64(12), o623–o625 CAS.
- Y. A. Chesalov, V. P. Baltakhinov, T. N. Drebushchak, E. V. Boldyreva, N. V. Chukanov and V. A. Drebushchak, J. Mol. Struct., 2008, 891(1), 75–86 CrossRef CAS.
- V. A. Drebushchak, T. N. Drebushchak, N. V. Chukanov and E. V. Boldyreva, J. Therm. Anal. Calorim., 2008, 93(2), 343–351 CrossRef CAS.
- T. N. Drebushchak, Y. A. Chesalov and E. V. Boldyreva, Acta Crystallogr., Sect. B: Struct. Sci., 2009, 65(6), 770–781 CAS.
- T. N. Drebushchak, V. A. Drebushchak and E. V. Boldyreva, Acta Crystallogr., Sect. B: Struct. Sci., 2011, 67(2), 163–176 CAS.
- H. Ueda, N. Nambu and T. Nagai, Chem. Pharm. Bull., 1984, 32(1), 244–250 CrossRef CAS.
- E. V. Boldyreva, V. Dmitriev and B. C. Hancock, Int. J. Pharm., 2006, 327(1), 51–57 CrossRef CAS PubMed.
- Y. V. Seryotkin, T. N. Drebushchak and E. V. Boldyreva, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, 69(1), 77–85 CAS.
- B. A. Zakharov, S. V. Goryainov and E. V. Boldyreva, CrystEngComm, 2016, 18(29), 5423–5428 RSC.
- B. A. Zakharov, Y. V. Seryotkin, N. A. Tumanov, D. Paliwoda, M. Hanfland, A. V. Kurnosov and E. V. Boldyreva, RSC Adv., 2016, 6(95), 92629–92637 RSC.
- T. N. Drebushchak, A. A. Ogienko and E. V. Boldyreva, CrystEngComm, 2011, 13(13), 4405–4410 RSC.
- I. A. Tumanov, A. F. Achkasov, S. A. Myz, E. V. Boldyreva and V. V. Boldyrev, Dokl. Chem., 2014, 457(2), 154–159 CrossRef CAS.
- A. A. L. Michalchuk, I. A. Tumanov, V. A. Drebushchak and E. V. Boldyreva, Faraday Discuss., 2014, 170, 311–335 RSC.
- I. A. Tumanov, A. F. Achkasov, E. V. Boldyreva and V. V. Boldyrev, CrystEngComm, 2011, 13(7), 2213–2216 RSC.
- A. A. L. Michalchuk, I. A. Tumanov and E. V. Boldyreva, CrystEngComm, 2013, 15(32), 6403–6412 RSC.
- I. A. Tumanov, A. F. Achkasov, E. V. Boldyreva and V. V. Boldyrev, Russ. J. Phys. Chem. A, 2012, 86(6), 1014–1017 CrossRef CAS.
- D. A. Rychkov, S. G. Arkhipov and E. V. Boldyreva, J. Appl. Crystallogr., 2014, 47(4), 1435–1442 CrossRef CAS.
- W. Ostwald, Z. Phys. Chem., 1897, 22(1), 289–330 CAS.
- J. Nývlt, Cryst. Res. Technol., 1995, 30(4), 443–449 CrossRef.
- T. Threlfall, Org. Process Res. Dev., 2003, 7(6), 1017–1027 CrossRef CAS.
- I. A. Tumanov, A. A. L. Michalchuk, A. A. Politov, E. V. Boldyreva and V. V. Boldyrev, CrystEngComm, 2017, 19(21), 2830–2835 RSC.
- A. V. Trask, N. Shan, W. D. S. Motherwell, W. Jones, S. Feng, R. B. H. Tan and K. J. Carpenter, Chem Commun., 2005, 880–882 RSC.
- D. Hasa, E. Miniussi and W. Jones, Cryst. Growth Des., 2016, 16, 4582–4588 CAS.
- F. Fischer, A. Heidrich, S. Greiser, S. Benemann, K. Rademann and F. Emmerling, Cryst. Growth Des., 2016, 16, 1701–1707 CAS.
- A. M. Belenguer, G. I. Lampronti, A. J. Cruz-Cabeza, C. A. Hunter and J. K. M. Sanders, Chem. Sci., 2016, 7, 6617–6627 RSC.
- D. Braga, S. L. Giaffreda, F. Grepioni, M. R. Chierotti, R. Gobetto, G. Palladino and M. Polito, CrystEngComm, 2007, 9(10), 879–881 RSC.
- G. A. Bowmaker, Chem. Commun, 2013, 49(4), 334–348 RSC.
- T. Friščić, S. L. Childs, S. A. Rizvi and W. Jones, CrystEngComm, 2009, 11(3), 418–426 RSC.
- C. Maheshwari, A. Jayasankar, N. A. Khan, G. E. Amidon and N. Rodríguez-Hornedo, CrystEngComm, 2009, 11(3), 493–500 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: X-ray diffraction patterns of samples crystallised under different conditions and obtained on different types of mechanical treatment. See DOI: 10.1039/c7ce02221b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |