DOI:
10.1039/C7CE01880K
(Paper)
CrystEngComm, 2018,
20, 796-806
Breaking the strong 1D growth habit to yield quasi-equiaxed REPO4 nanocrystals (RE = La–Dy) via solvothermal reaction and investigation of photoluminescence†
Received
30th October 2017
, Accepted 3rd January 2018
First published on 3rd January 2018
Abstract
Quasi-equiaxed REPO4 (RE = La–Dy) nanocrystals of either monoclinic (m-) or hexagonal (h-) structure, depending on the type of RE3+, have been successfully synthesized through solvothermal reaction in the presence of ethylene glycol (EG). Detailed characterization of the products was achieved by the combined XRD, FE-SEM, TEM, FTIR, TG/DTA, PLE/PL, and fluorescence decay techniques. The intrinsically strong 1D growth of the phosphates was gradually suppressed through increasing the solution pH and EG dosage, and the formation mechanism was elucidated with h-GdPO4 as a representative. The differences in the crystal structure, IR response and crystal size of the as-synthesized REPO4 crystals were interpreted in detail. It was shown that the phase purity and crystal morphology of h-GdPO4 can be well retained up to 700 °C, followed by a complete transition from the hexagonal to monoclinic phase at 900 °C. The photoluminescence properties of Dy3+-doped h-GdPO4 nanocrystals, including excitation, emission and fluorescence decay, were thoroughly investigated, and almost pure white-light luminescence was attained with similarly strong yellow (573 nm, 4F9/2 → 6H13/2) and blue (478 nm, 4F9/2 → 6H15/2) emissions of Dy3+ under excitation of the host Gd3+ ions at ∼274 nm.
1. Introduction
Rare-earth (RE) orthophosphates (REPO4) are attracting wide attention due to their superior advantages such as high thermal stability (melting point ∼2300 °C),1 extremely low water solubility (solubility product ∼10−25 to 10−27),2 and high concentration of doped lasing ions (∼1.8 × 1021 ions per cm3).3 In addition, the unique electronic, optical, magnetic, and catalytic properties of RE elements endow REPO4 with multiple functionalities.4,5 As inorganic nanocrystals frequently exhibit many size- and morphology-associated novel properties,6 morphology controllable synthesis of REPO4 has attracted extensive interest, and it embodied great significance in many domains such as lighting, biolabeling, phototherapy, and optical imaging.7 Till now, REPO4 nanostructures with a variety of configurations have been successfully fabricated by a number of techniques, including sol–gel,8 the sacrificial template method,9 microemulsion synthesis,10 microwave-assisted crystallization,11 and so on. Among the developed synthetic strategies, hydro-/solvothermal reaction showed outstanding capacity since it not only directly produces well-crystallized products within a short period of reaction but also allows versatile interior reaction conditions.12,13 Fang et al., for example, hydrothermally synthesized REPO4 nanowires (RE = La–Dy) and ill-shaped nanoparticles (RE = Ho–Lu and Y) without the use of any additive.14 Through applying oleic acid (OA) as a surfactant, Huo et al. solvothermally synthesized REPO4 crystals with multiple morphologies of nanowires, nanorods, and nanoparticles with cuboid and hexagonal shapes in an ethanol/water mixture.15
It is well documented that REPO4 may adopt three types of crystal structures depending on the effective ionic radius of RE. Generally, larger La–Tb cations prefer to coordinate with 9 oxygen ions (CN = 9; CN: coordination number) to crystallize in the stable monoclinic phase (m-REPO4) while smaller Dy–Lu and Y cations tend to crystallize in the stable tetragonal phase (t-REPO4, CN = 8).16,17 Besides, the metastable hexagonal phase (h-REPO4, CN = 8) may be adopted by the whole lanthanide spectrum and Y under appropriate conditions.14,15 Owing to the relatively lower activation energy of growth along the [001] direction (c-axis), m-/h-REPO4 crystals always present strong 1D growth preference under wet chemical synthesis conditions.15,18,19 Though a lower solution pH value sometimes proved to be beneficial to 1D growth,20–22 a systematic account of the role of pH in the phase/shape evolution of REPO4 can hardly be found.
The intrinsic thermodynamic growth pattern of a crystal is not always likely to be the crucial factor in determining the shape evolution. A variety of strategies, such as manipulation of monomer concentration,23,24 the use of appropriate capping agents,25 and control over the anion-exchange rate,7 have been extensively examined and found to be useful in regulating the growth habit of crystals under hydrothermal conditions. It is worth mentioning that the solubility of reaction products and the reactivity and diffusion behaviors of reactants significantly vary with the type of reaction medium (solvent).26 Accordingly, it is possible to exert precise control over the nucleation and growth patterns of crystals for various materials through solvothermal reaction in an appropriate solvent. You et al., for example, synthesized YBO3 and YVO4 microstructures via an EG-assisted hydrothermal route, and the effects of the reaction time, pH value, and amount of ethylene glycol on the properties of the products were studied.27,28 As for REPO4, Becerro et al. synthesized intersecting lance-shaped m-GdPO4 nanoparticles with an ethylene glycol/water mixture as the solvent.29 Despite these successes, breaking the stubborn 1D growth habit of m-/h-REPO4 to produce equiaxed nanocrystals via solvothermal reaction is still hard to reach.
Aside from Eu3+, Dy3+ is a well-known activator frequently used to probe the residential crystal environment of activator because of its hypersensitive yellow emission (4F9/2 → 6H13/2 transition, ∼575 nm).30 Furthermore, suitably adjusting the intensity ratio of its yellow to blue (4F9/2 → 6H15/2, ∼480 nm) emissions may realize white luminescence of a phosphor solely doped with Dy3+. By applying solvothermal reaction in an EG/H2O mixture and controlling precisely the pH value, the significant 1D growth of REPO4 (RE = La–Dy) was blocked and quasi-equiaxed nanocrystals of m-/h-REPO4 (RE = La–Dy) have been successfully synthesized in this work. Almost pure-white emission was also realized through Dy3+ doping of h-GdPO4. In the present study, the morphology evolution of the nanocrystals was thoroughly elaborated by varying the pH value and EG content with h-GdPO4 as a representative, and the effects of lanthanide contraction and REPO4 solubility on the crystal structure and morphology of REPO4 were elucidated. Furthermore, the thermal behaviors of the as-synthesized h-GdPO4 nanocrystals and the phase/morphology evolution upon calcination were also investigated in detail.
2. Experimental
2.1. Solvothermal synthesis of REPO4 crystals
The starting materials (Kanto Chemical Co., Inc., Tokyo, Japan) are RE(NO3)3·6H2O (RE = La–Dy, >99.99% pure), HNO3 (ultrahigh purity), sodium hydroxide (NaOH, >99% pure), and ethylene glycol (EG, >99.5% pure). Milli-Q filtered water (resistivity ∼18.2 MΩ cm) was used throughout the experiment. In a typical synthesis, certain amounts of EG and H2O were mixed together to make a 60 mL transparent solvent, to which 1 mmol of RE3+ was then added. After vigorous stirring for 15 min, 1 mmol NaH2PO4 was added and the pH was adjusted to a certain value with NaOH/HNO3. The resultant mixture was constantly stirred for 30 min before being transferred to a Teflon-lined stainless steel autoclave of 100 mL capacity. The autoclave was tightly sealed and was put into an electric oven preheated to 200 °C. After 24 h of reaction, the autoclave was left to cool naturally to room temperature and the hydrothermal product was collected via centrifugation, followed by washing with water three times and ethanol once and air drying at 70 °C for 24 h. Calcination of the hydrothermal product was carried out in stagnant air, using a heating rate of 10 °C min−1 and a duration time of 2 h. After annealing, the sample was cooled to 300 °C at 10 °C min−1 and then naturally cooled to room temperature.
2.2. Characterization
Phase identification was performed via X-ray diffractometry (XRD; Model RINT2200, Rigaku, Tokyo, Japan) at 40 kV/40 mA, using nickel-filtered Cu-Kα radiation (0.15406 nm) and a scanning speed of 4°/2θ per minute. The product morphology was analyzed by field emission scanning electron microscopy (FE-SEM, Model S-4800, Hitachi, Tokyo, Japan) operating at 10 kV and transmission electron microscopy (TEM, JEM-2100F, JEOL, Tokyo) at 200 kV. Fourier transform infrared spectroscopy (FT-IR; Model 4200, JASCO, Tokyo) was performed by the standard KBr pellet method. Thermogravimetry/differential thermal analysis (TG/DTA; Model STA449F3, Jupiter, NETZSCH, Germany) of the as-synthesized h-GdPO4 was conducted under flowing simulated air (50 mL min−1) up to 1000 °C at a heating rate of 10 °C min−1. Photoluminescence properties were analyzed at room temperature with an FP-8500 fluorospectrophotometer (JASCO), which has a spectral resolution of ±1.5 nm. Fluorescence decay at room temperature was measured by a time-correlated single photon counting system (TCSPC), using a 370 nm nanoLED as the excitation source (1.2 ns pulse duration, Horiba Jobin Yvon, FluoroCube, Tokyo).
3. Results and discussion
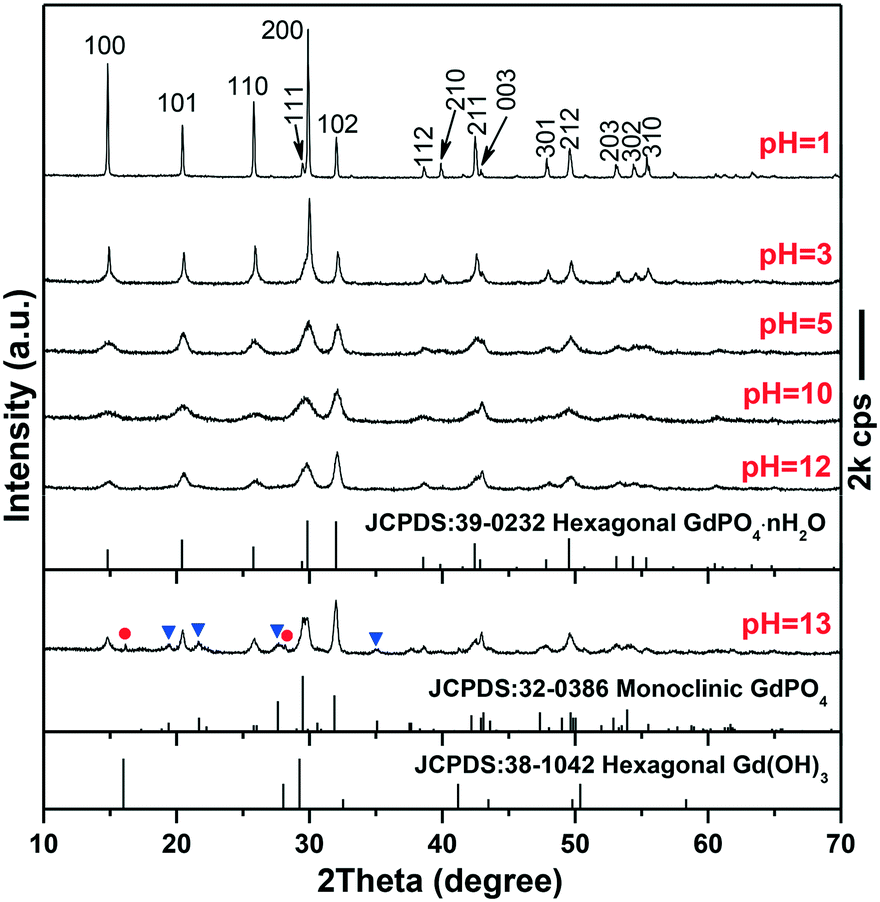
3.1. The effects of pH on the structure and morphology of GdPO4
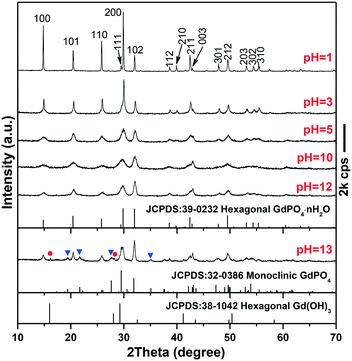
The phase/morphology evolution of GdPO4 at 200 °C of hydrothermal reaction was firstly investigated against solution pH (Fig. 1). It is seen that hexagonal structured GdPO4 (h-GdPO4; JCPDS no. 39-0232, space group P3121) was obtained at the lowest pH of 1,31 whose phase purity well retained up to pH = 12 though the crystallinity was gradually degraded with increasing pH. Reaction at a higher pH value of 13 led to co-crystallization of small amounts of monoclinic GdPO4 (m-GdPO4; JCPDS no. 32-0386) and hexagonal Gd(OH)3 (JCPDS no. 38-1042) impurities, as denoted by the blue triangles and red dots, respectively. The morphology variation with pH is displayed in Fig. 2, where it is seen that the h-GdPO4 crystals all present 1D morphologies of varying aspect ratios. Fig. 2a and b show that the pH = 1 sample consists of microwires with lengths of ∼2–6 μm and diameters of ∼40–400 nm. The SAED analysis (the inset in Fig. 2b) of a single wire yields a set of well-defined diffraction spots that correspond to the (001), (100), and (101) planes of h-GdPO4, indicating that each of the microwires is single crystalline and well crystallized. The HR-TEM analysis of a single wire (Fig. 2c) reveals an interplanar distance of 0.60 ± 0.01 nm that corresponds to the (100) plane of h-GdPO4 (d100 = 0.598 nm), which suggests the preferred growth of the wire along the [001] direction (arrowed). In addition, the (101) and (001) fringes can also be observed with spacings of 0.43 ± 0.01 and 0.63 ± 0.01 nm, respectively (d101 = 0.435 nm, d001 = 0.633). Although the pH = 3 sample is a mixture of micro- and nanowires (Fig. 2d), the pH = 5 product exclusively contains nanowires with lengths of ∼90–600 nm and diameters of ∼15–20 nm (Fig. 2e). Further increasing the pH value to 10 or 12 did not bring about any obvious change to the wire diameter but significantly shortened the wire length to ∼70–200 nm (Fig. 2f–h). The SAED rings (the inset in Fig. 2h) taken from the nanowires can be assigned to the {100}, {101}, and {200} planes of h-GdPO4, and the HR-TEM lattice image (Fig. 2i) clearly reveals that the nanowires also grew up along the [001] direction though they are much thinner and shorter than the microwires.
 |
| | Fig. 1 XRD patterns of the samples synthesized with different pH values indicated in the figure. The blue triangles and red dots in the pattern of the pH = 13 sample denote monoclinic GdPO4 and hexagonal Gd(OH)3, respectively. | |
 |
| | Fig. 2 FE-SEM (a and d–g) and TEM (b and h) micrographs showing the morphologies of the h-GdPO4 products synthesized with different pH values of 1 (a and b), 3 (d), 5 (e), 10 (f), and 12 (g and h). The insets in (b) and (h) are the selected area electron diffraction (SAED) patterns. Parts (c) and (i) are the HR-TEM lattice images of the pH = 1 and 12 samples, respectively. | |
It is known that the liberation of PO43− from NaH2PO4 is highly dependent on solution pH. At the very low pH value of 1, the PO43− anions would be almost totally captured by H+ to form H3PO4, which prohibited GdPO4 nucleation, and thus, a clear solution of the reactants was observed before the hydrothermal reaction. As the temperature increases, a small amount of PO43− would be liberated from H3PO4 to induce GdPO4 nucleation. Under this circumstance, the low nucleation density and hence the high concentrations of remaining solutes facilitated the full growth of GdPO4 crystals, resulting in the microwires with large lengths and diameters (Fig. 2a and b). Increasing the pH value to 3 largely promoted the liberation of PO43− from the hydrogen phosphate species (H2PO4− and HPO42−), and thus, the precipitation of h-GdPO4 took place prior to hydrothermal reaction. Due to the low activation energy at room temperature, the resultant h-GdPO4 was poorly crystallized and appeared as severely aggregated spiculate particles, as revealed by the broadened XRD peaks and SEM observation (Fig. S1 and its inset†). The room-temperature precipitation would lower the solute concentration, which limited the crystallite growth during the hydrothermal reaction, and thus, averagely thinner microwires were yielded (Fig. 2d). The solutes tend to completely precipitate from the solution generating poorly crystallized h-GdPO4 at pH ≥5, for which h-GdPO4 crystals with smaller diameters and lengths were produced by hydrothermal reaction (Fig. 2e–g). It is noteworthy that, although H3PO4 undergoes incomplete dissociation under acidic conditions according to H3PO4 ↔ H+ + H2PO4− ↔ 2H+ + HPO42− ↔ 3H+ + PO43−, the stronger ability of PO43− than H2PO4− and HPO42− to coordinate with RE3+ leads to the preferential formation of REPO4 nuclei.32 This would in turn right-shift the above equilibrium and promote the dissociation of H3PO4/H2PO4−/HPO42− species until Gd3+ is completely consumed. When the pH value reached 13, the large OH− concentration led to partial crystallization of hexagonal Gd(OH)3 with h-GdPO4. Owing to the stronger ability of PO43− than OH− to coordinate with RE3+, however, some Gd(OH)3 would be transformed into m-GdPO4, rather than h-GdPO4, upon hydrothermal reaction, since in the two phases the Gd3+ ions are similarly 9-fold coordinated.33,34 This accounts for the observed phase mixture of h-Gd(OH)3, m-GdPO4, and h-GdPO4.
The intensity ratio R of (200) to (102) diffraction peaks as a function of solution pH is depicted in Fig. S2.† Combined with the results of SEM analysis, it can be recognized that the R value decreased from ∼3.51 at pH = 1 to ∼2.43 at pH = 3 along with the decrease in the overall diameter of the microwires (no obvious change in wire length). The R value then rapidly decreased to ∼1.17 at pH = 5 with the decrease in the amount of the microwires and the significant appearance of thinner and shorter nanowires (Fig. 2e), followed by a slow decrease to ∼0.73 at pH = 12 along with shortening of the nanowires. It can therefore be deduced that the shrinking diameter, instead of length, of the wires contributed more to the decreasing R value. This is understandable in view that decreasing the wire diameter directly reduces the stacking thickness of {200} facets (Fig. 2c) and lowers the diffraction intensity of the facets.
3.2. The effects of EG on the phase and morphology of GdPO4
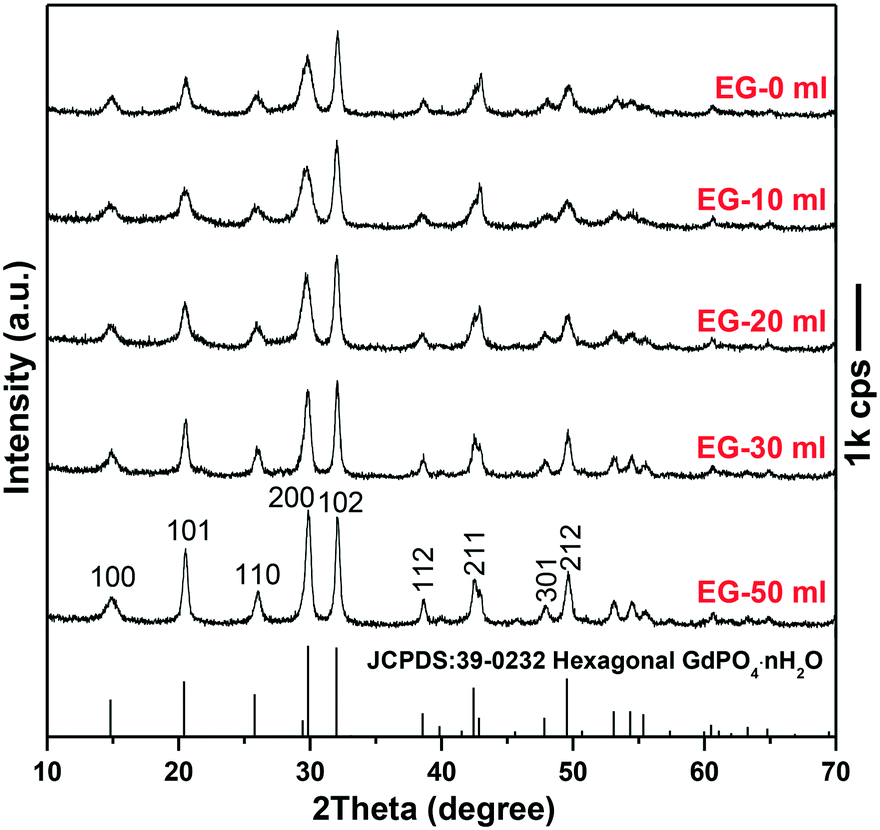
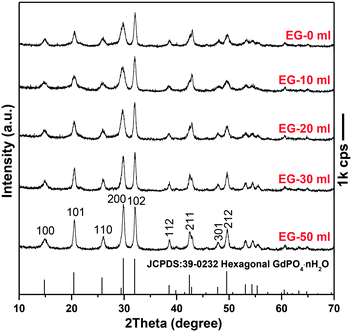
Since the 1D growth of h-GdPO4 crystals was significantly restricted under alkaline hydrothermal conditions (Fig. 2), the pH value of 12 was chosen to investigate the effects of EG on the phase composition and morphology of GdPO4 crystals. It is seen from Fig. 3 that the addition of EG did not change the phase purity of h-GdPO4 but the intensity ratio (R) of the (200) to (102) diffraction peaks gradually increased from ∼0.71 ± 0.02 for EG = 0 and 10 mL to ∼1.05 for EG = 50 mL (∼70 vol% of the total solvent), as depicted in Fig. S3.† The SEM images of the EG-assisted products are displayed in Fig. 4a–d, where it is seen that increasing EG addition gradually shortened the length of the nanowires and finally led to the crystallization of quasi-equiaxed h-GdPO4 nanocrystals at EG = 30 and 50 mL (Fig. 4c and d). The TEM observation (Fig. 4e and the inset) found that the nanocrystals possess hexagonal top/bottom surfaces, corresponding well with the hexagonal crystal structure. HR-TEM analysis (Fig. 4f) of a single nanoparticle yields a set of well-resolved lattice fringes that are attributable to the (101) plane of h-GdPO4. The lattice fringes spanned the whole particle, indicating that each individual nanocrystal is single crystalline. The “diameter” (shorter side) of the products were measured and labeled in Fig. 4a–d, and the aspect ratio of the quasi-equiaxed h-GdPO4 nanocrystals was assayed to be below 2. It was found that EG addition slightly yet steadily increased the crystallite diameter from ∼10–20 nm (Fig. 2g, EG = 0 mL, R = 0.73) to 30–37 nm (Fig. 4d, EG = 50 mL, R = 1.05), which may account for the increased R value and the gradually sharpened (200) diffraction (Fig. 3) since the (200) diffraction is more sensitive to the particle diameter as inferred from Fig. 2c. It is also interesting to notice that the quasi-equiaxed h-GdPO4 nanocrystals grew from ∼25–35 to ∼30–37 nm with increasing EG content from 30 to 50 mL (Fig. 4c and d).
 |
| | Fig. 3 XRD patterns of the h-GdPO4 samples synthesized with different EG contents. | |
 |
| | Fig. 4 FE-SEM (a–d) and TEM (e) images showing the morphologies of the h-GdPO4 samples synthesized with different EG contents of 10 (a), 20 (b), 30 (c), and 50 mL (d and e). The insets in (e) show the hexagonal top/bottom surface of a nanocrystal. Part (f) and its inset are the HR-TEM lattice image and SAED pattern of the EG = 50 mL sample, respectively. | |
The observed shape evolution of h-GdPO4 crystals can be explained by the selective absorption of EG on specific crystal surfaces and the nucleation kinetics governed by EG. Since the 1D growth was gradually inhibited, it can be deduced that the EG molecules selectively cap the {001} planes of h-GdPO4 to hinder the crystal growth along the [001] direction.27,28,35 The intrinsically preferred [001] growth of h-GdPO4 was almost completely suppressed when the EG volume was 30 mL, and thus, the quasi-equiaxed nanocrystals were obtained. Since the concentrations of Gd and phosphate sources were constant in this work, the significantly prohibited 1D growth by EG capping was expected to yield crystals of substantially larger diameters, but only a slight diameter increase was observed. This could be due to the increased nucleation density as discussed below. For chemical precipitation, the final particle size is known to be closely related to the equilibrium concentration of solutes in saturated solution (the solubility of precipitate), which can be estimated from the following equation proposed by Chen et al.:36
| | | Cs ≈ exp{−(z+z−e2)/[4πε0εrkT(rz+ + rz−)]} | (1) |
where
rz+ and
rz− are the radii of ions charged by
z+ and
z−, respectively;
ε0 is the vacuum permittivity and
εr is the relative dielectric constant of the solvent;
k is the Boltzmann's constant, and
T is the temperature in Kelvin. Owing to the smaller dielectric constant of EG (37.7) than water (78.5),
37 the solubility (
Cs) of GdPO
4 would successively decrease with increasing EG addition, thus leading to gradually higher supersaturation for nucleation. On the other hand, the free energy of critical nucleus formation, Δ
Gcrit, can be given by:
38where
γ is the liquid/solid interfacial tension and
rc is the critical nucleus radius. EG has a lower surface energy (48.4 N m
−1) than water (72.8 N m
−1),
37 and hence, its presence would reduce the energy barrier for stable nuclei formation through lowering the
γ term in
eqn (2). The above two factors may therefore raise the nucleation density of GdPO
4, and as a result, only slightly larger crystal diameter was observed despite the significantly prohibited 1D crystal growth. It should also be noted that a high enough EG content may hinder PO
43− dissociation from NaH
2PO
4 due to the poor accessibility of water molecules, which would in turn lower the nucleation density of GdPO
4. This may account for the size increment of the quasi-equiaxed nanocrystals upon raising the EG content from 30 to 50 mL.
3.3. Extension to the synthesis of other REPO4 nanocrystals
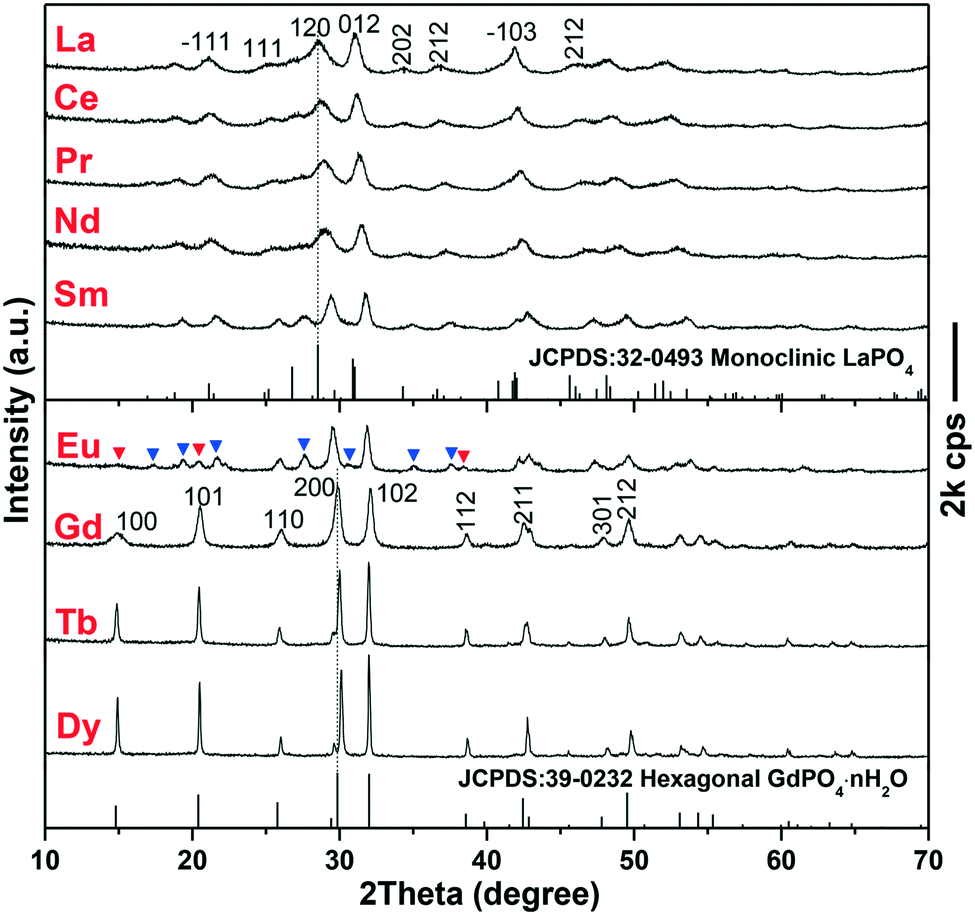
The EG-assisted approach developed for h-GdPO4 nanocrystals was implemented in the synthesis of other REPO4 (RE = La–Eu, Tb, and Dy) crystals that similarly present intrinsic 1D growth preference. XRD analysis of the products (Fig. 5) shows that the phosphates of larger La3+–Sm3+ cations crystallized in the monoclinic system while those of smaller Tb3+ and Dy3+, similar to Gd3+, crystallized as hexagonal phases. The Eu product is a mixture of the above two phases, as denoted by the blue and red triangles in Fig. 5, which may suggest that Eu is the boundary element for phase selection. The above results can be attributed to lanthanide contraction, that is, larger RE cations show stronger ability to coordinate with more oxygen ions and thus favor m-REPO4 crystallization (CN = 9 for m-REPO4 and CN = 8 for h- and t-REPO4).14–18 The diffraction peaks tend to shift to the high-angle region from m-LaPO4 to m-SmPO4 and from h-GdPO4 to h-DyPO4, and the cell parameters of both the m- and h-REPO4 crystals (derived with MDI Jade 5.0 software) linearly decrease with decreasing RE3+ size (Fig. S4†), conforming well to lanthanide contraction. Analysis of the diffraction peak broadening with the Scherrer equation yielded average crystallite sizes of ∼9 ± 3 nm for m-REPO4 (RE = La–Nd), ∼15 nm for m-SmPO4, ∼36 nm for h-GdPO4, and ∼60 nm for h-TbPO4.
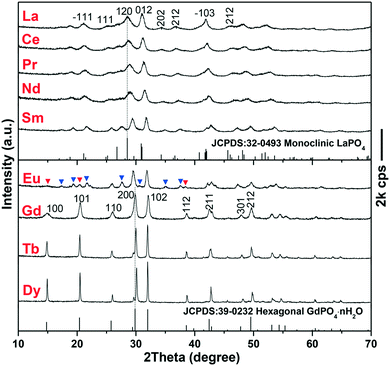 |
| | Fig. 5 XRD patterns of the REPO4 products obtained via solvothermal reaction at 200 °C, pH = 12, and EG = 50 mL for 24 h. The blue and red triangles in the XRD pattern of the Eu product denote the monoclinic and hexagonal phases, respectively. | |
The morphologies of m-REPO4 (RE = La–Nd) were analyzed via SEM and TEM with m-LaPO4 as an example (Fig. 6a and b). It was found that m-LaPO4 consists of nanoparticles with extremely tiny sizes of ∼10 nm, which is in agreement with the crystallite size assayed via XRD. The SAED analysis (the inset in Fig. 6b) clearly reveals the diffraction rings assignable to the {200} and {012} planes of m-LaPO4. The HR-TEM lattice image (Fig. 6c) exhibits that the nanoparticles are structurally uniform, with the interplanar distance of 0.32 ± 0.01 nm corresponding well to the (120) plane (d120 = 0.313 nm). The m-SmPO4 sample also consists of quasi-equiaxed particles/crystallites, similar to m-LaPO4, but the particle size increased to ∼15–20 nm (Fig. 6d and its inset). The h-TbPO4 and h-DyPO4 products (Fig. 6e and f) consist of elongated particles with a diameter/length of ∼40/80 and 60/200 nm, respectively. Firsching et al. reported that the solubility of REPO4 slowly increases from La to Eu and then rapidly rises to the maximum values of Tb and Dy,2 which well explains the steadily increasing crystal size from La–Dy since higher solubility would lower the nucleation density and thus lead to a larger final crystal size. The elongated phosphate crystals observed in Tb and Dy, particularly Dy, may suggest that the 1D growth preference becomes stronger with decreasing RE3+size. In spite of this, the h-TbPO4 and h-DyPO4 particles have small aspect ratios of only ∼2–4, which implies that the strong 1D growth tendency has indeed been effectively inhibited by the synthetic approach employed in this work.
 |
| | Fig. 6 FE-SEM (a and d–f) and TEM (b) images showing the morphologies of the m-/h-REPO4 products, where RE = La (a and b), Sm (d), Tb (e) and Dy (f). Part (c) is the HR-TEM lattice image of several m-LaPO4 nanocrystals. The insets in (b) and (d) are the corresponding SAED pattern and high magnification SEM micrograph, respectively. | |
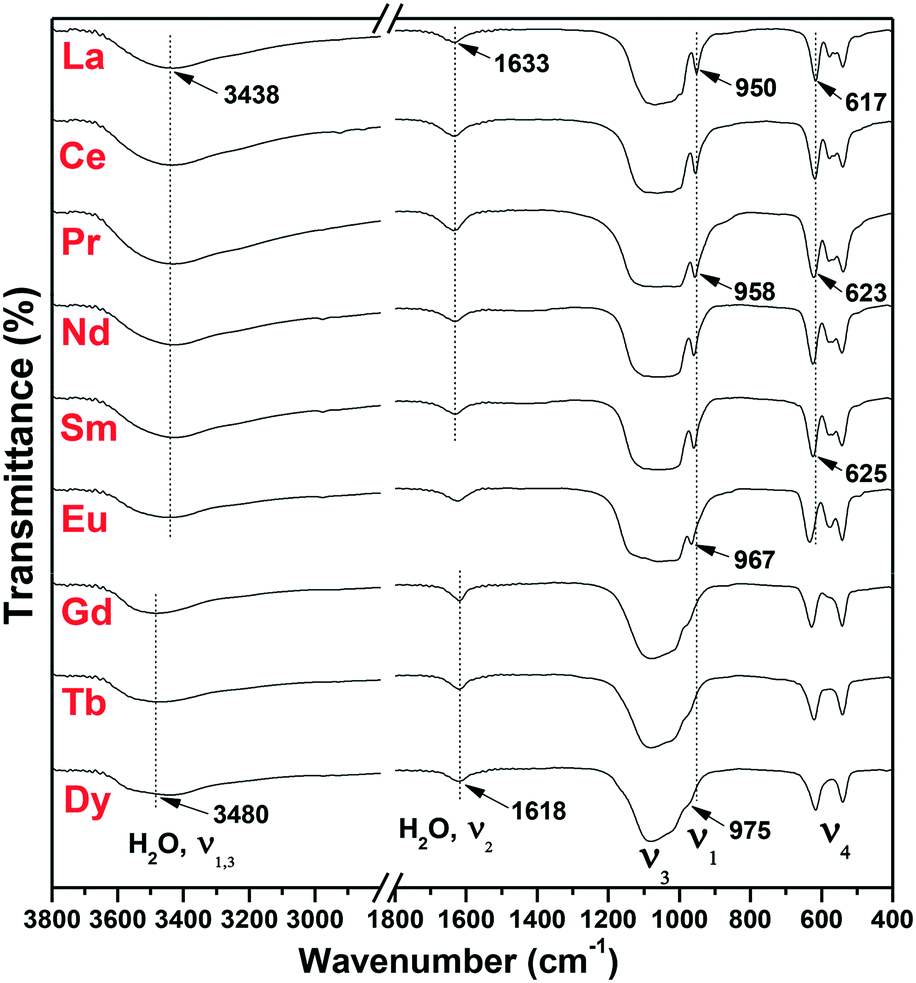
The FTIR analysis of the as-synthesized m-/h-REPO4 products is displayed in Fig. 7. For m-REPO4 (La–Sm), the PO43− ions present a broad vibration band ranging from ∼500 to 1200 cm−1, with a sharp peak at ∼960 cm−1 attributable to the P–O symmetric stretching vibration (ν1), the broad absorption at ∼980–1200 cm−1 to the P–O antisymmetric stretching (ν3), and the splits in the ∼500–700 cm−1 region to the P–O bending mode (ν4).39 The absorptions at ∼3438 and 1633 cm−1 can be assigned to the O–H stretching vibrations (ν1 and ν3) and H–O–H bending mode (ν2) of water,40 respectively. Compared with m-REPO4, h-REPO4 (Gd–Dy) exhibited lower resolutions of ν1 and ν4 PO43− vibrations owing to the higher symmetry of PO43− in h-REPO4 (C2) than that in m-REPO4 (C1).41 The ν1 vibration peak of PO43− was found to shift toward the higher energy (from ∼950 to 975 cm−1) with decreasing RE3+ size, owing to the stronger polarizing power of smaller RE3+.42 The vibration frequency increases towards smaller RE3+ for the ν4 component located at ∼620 cm−1, as also observed by Ross et al., though the exact reason is yet unknown.39 It is interestingly noticed that the O–H stretching vibration of H2O has larger frequency in h-REPO4 than in m-REPO4 while an opposite trend is seen for the H–O–H bending mode. There are two reasons for such a phenomenon: (1) the hydration water in h-REPO4 has stronger bonding force with the crystal framework than the absorbed water in m-REPO4, and thus the stretching vibration needs higher energy, and (2) the hydration effect may enable more lone-pair electrons from the 3a1 orbital of water to take part in chemical bonding in h-REPO4, which enlarges the H–O–H bond angle and thus lowers the frequency of bending vibration.43
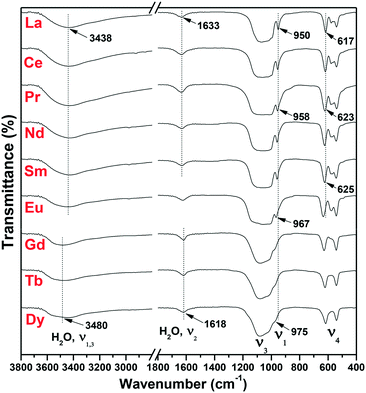 |
| | Fig. 7 FTIR spectra of the REPO4 products. | |
3.4. Thermal behavior of the as-synthesized h-GdPO4 nanocrystals
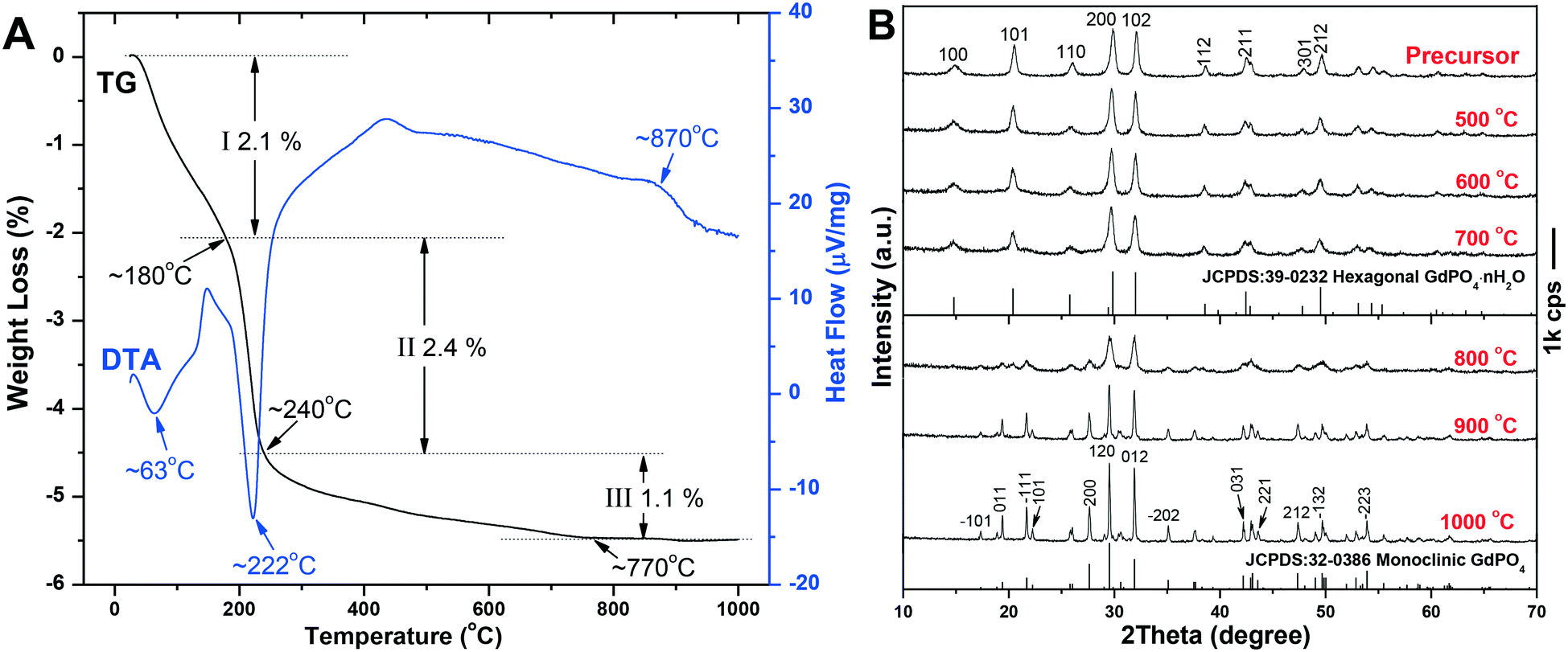
The TG/DTA analysis of the h-GdPO4 nanocrystals (EG = 50 mL) is depicted in Fig. 8A, where it is seen that the sample loses weight via three separate steps upon heating. The first weight loss of ∼2.1% up to ∼180 °C, accompanied by an endotherm centered at ∼63 °C, is largely due to the elimination of adsorbed water. The rapid loss of ∼2.4% in the narrow temperature range of ∼180–240 °C is ascribed to the removal of water bound to PO43− through hydrogen bonding, which produces a sharp endotherm at ∼222 °C. The subsequent weight loss of ∼1.1%, taking place in the wide temperature range of ∼240–770 °C, can be attributed to the removal of hydration water from the crystal bulk. The number of hydration (n) in each h-GdPO4·nH2O formula can thus be determined to be ∼0.16. The exotherm located at ∼870 °C, without the occurrence of any weight loss, is associated with the h- to m-GdPO4 phase transition as evidenced by the XRD analysis results (Fig. 8B). It is clear that the hexagonal structure can be well retained up to 700 °C, followed by a complete transition to m-GdPO4 at 900 °C (Fig. 8B). Rietveld refinement of the XRD patterns of the 700, 800, and 900 °C products yielded well acceptable weighted profile R-factors (Rwp) in each case (Fig. S5†). It is also clear that the 700 °C (Fig. S5a†) and 900 °C (Fig. S5c†) samples are of pure h- and m-GdPO4 phase, respectively, while the 800 °C one (Fig. S5b†) is a mixture of the two phases (m-GdPO4 content: ∼93.65 wt%). The results thus further confirmed the phase transition observed in Fig. 8. Calcination at a higher temperature of 1000 °C did not alter the monoclinic crystal structure but significantly enhanced the crystallinity of the product. Analysis of the XRD peak broadening with the Jade 5.0 software yielded crystallite sizes of ∼26 and 79 nm for the samples calcined at 700 and 900 °C, respectively. FE-SEM observation (Fig. 9) found slight aggregation and deformation for the particles calcined at 700 °C (Fig. 9c), since the mass transfer via diffusion would bind the neighboring particles together and deform them. Calcination at 900 °C led to severe aggregation and deformation of the particles (Fig. 9d), because of the significant inter-particle sintering, crystallite growth, and re-constructive type hexagonal to monoclinic phase transition.
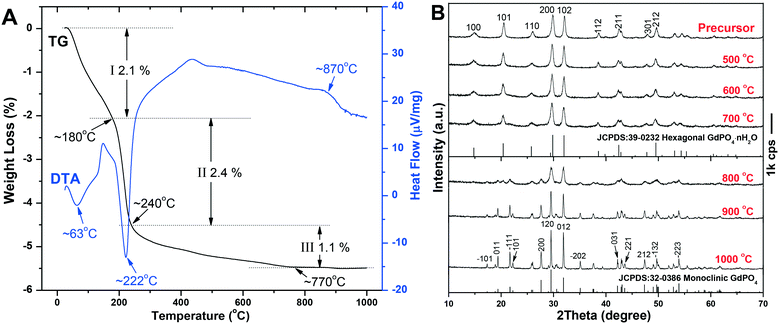 |
| | Fig. 8 TG-DTA profiles (A) and the temperature dependent phase evolution (B) of h-GdPO4. | |
 |
| | Fig. 9 FE-SEM images showing the morphologies of the h-GdPO4 products (EG = 50 mL) calcined at temperatures of 500 (a), 600 (b), 700 (c), and 900 °C (d). | |
3.5. Luminescence of Dy3+ in h-GdPO4 nanocrystals
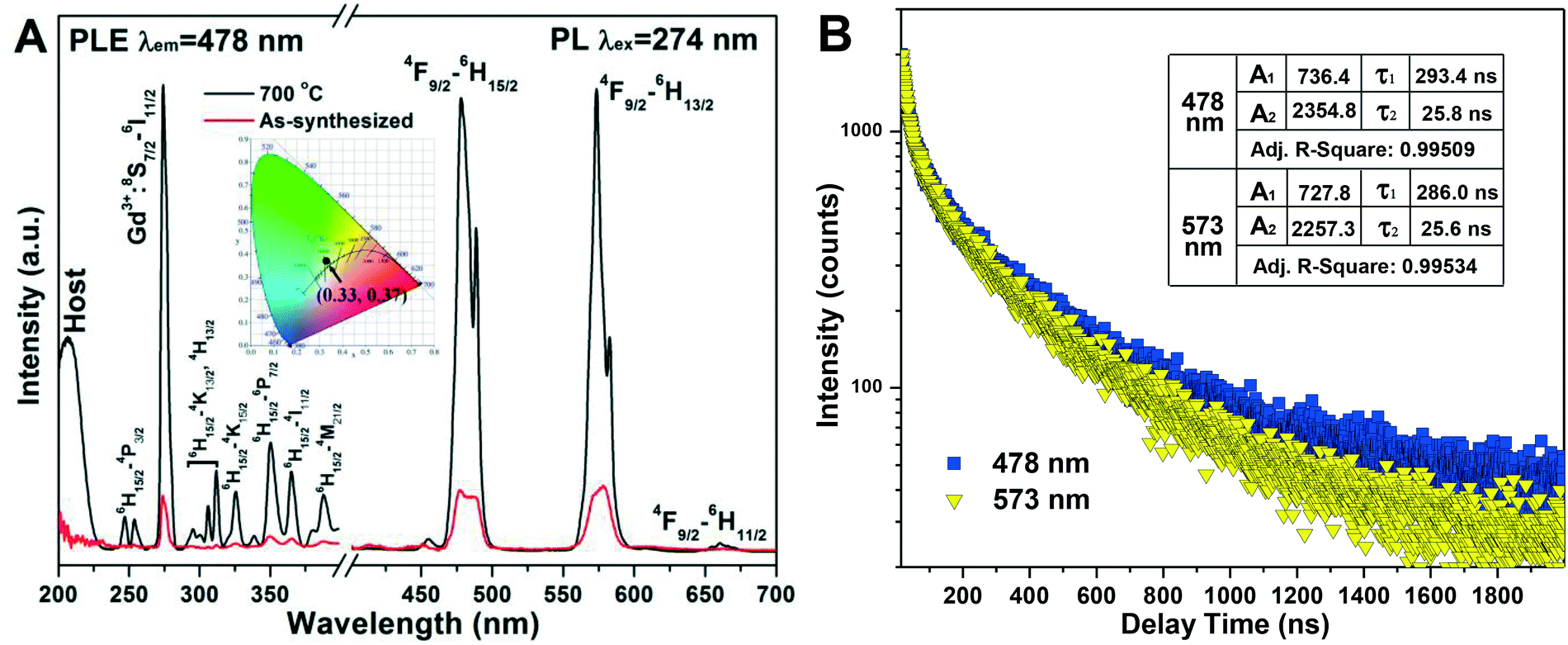
Fig. 10A shows the photoluminescence excitation (PLE) and photoluminescence (PL) spectra of h-(Gd0.95Dy0.05)PO4 (synthesized with EG = 50 mL) before and after 700 °C calcination, where it is seen that calcination raised both the excitation and emission intensities by a factor of ∼750%. Such drastic improvements are largely due to the almost complete elimination of water molecules, which are well known to cause serious luminescence quenching,9,44 from the surface and crystal interior of h-GdPO4, as demonstrated by the TG result (Fig. 8A). The PLE spectra (λem = 478 nm) exhibit several sharp peaks in the ∼230–400 nm region which include the 8S7/2 → 6I11/2 transition of Gd3+ at ∼274 nm and the weak f–f transitions of Dy3+ within the 4f9 configuration,45 as assigned in the figure. The peak at ∼210 nm is due to host absorption. The 8S7/2 → 6I11/2 Gd3+ transition appears to be significantly stronger than any other excitation band, implying an efficient Gd3+ → Dy3+ energy transfer. Under UV excitation at 274 nm, two predominant emission peaks were observed to arise from the 4F9/2 → 6H15/2 and 4F9/2 → 6H13/2 transitions of Dy3+ at 478 nm (blue) and 573 nm (yellow), respectively. Hardly perceptible is the following line at 660 nm associated with the 4F9/2 → 6H11/2 Dy3+ transition.46 The energy transfer from Gd3+ to Dy3+ is schematically displayed in Fig. S6.† Exciting the h-(Gd0.95Dy0.05)PO4 phosphor at 274 nm would raise Gd3+ electrons from the 8S7/2 ground state to the 6I11/2 energy level, followed by relaxation to the 6P7/2 state. Resonant energy transfer from Gd3+ to Dy3+ may then take place upon back jumping of the 6P7/2 Gd3+ electrons, through which the lowest-lying 6H15/2 ground-state electrons of the Dy3+ activator can be excited to the 4H13/2/4K13/2 energy level. The 4K13/2/4H13/2 Dy3+ electrons would relax to the 4F9/2 level in a non-radiative way, followed by radiative relaxation to the 6H15/2, 6H13/2, and 6H11/2 ground states to produce the observed blue (strong), yellow (strong), and red (very weak) emissions.
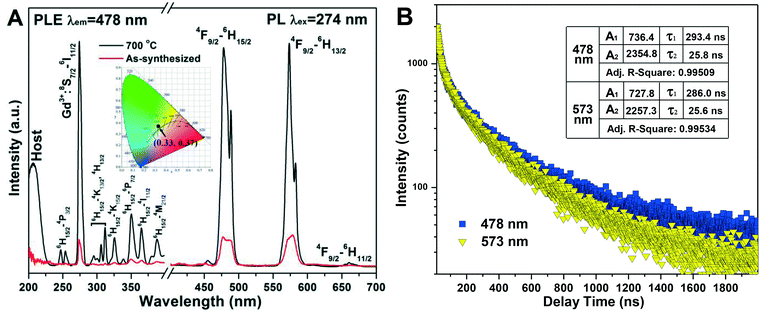 |
| | Fig. 10 Photoluminescence excitation (PLE) and emission (PL) spectra of the as-synthesized (EG = 50 mL) and 700 °C calcined h-(Gd0.95Dy0.05)PO4 (A) and fluorescence decay kinetics for the 478 and 573 nm emissions of the 700 °C calcined h-(Gd0.95Dy0.05)PO4 (B). The inset in (A) is the CIE chromaticity diagram of the 700 °C calcined h-(Gd0.95Dy0.05)PO4 phosphor. | |
The 4F9/2 → 6H13/2 emission is a forced electric dipole transition, whose intensity is hyper-sensitive to the crystal field around Dy3+, while the 4F9/2 → 6H15/2 magnetic dipole transition is not. According to the Judd–Ofelt parity law,47,48 the electric dipole transition is allowed only under the condition that the Dy3+ ions occupy a low symmetry site without an inversion center.49 In h-GdPO4, Dy3+ is expected to replace Gd3+ to inherit the centrosymmetric D2 point symmetry. The observed 4F9/2 → 6H13/2 transition is, however, as strong as the 4F9/2 → 6H15/2 transition. This can be attributed to the high surface area of the nanocrystals arising from their very limited crystallite size (∼40 nm), which allows some of the Dy3+ ions to be located at or near crystallite surfaces. The loss of point symmetry of these surface region Dy3+ enhances 4F9/2 → 6H13/2 while lowers the 4F9/2 → 6H15/2 emission at the same time. The h-(Gd0.95Dy0.05)PO4 phosphor calcined at 700 °C was calculated to obtain the Commission international de l'éclairage (CIE) chromaticity coordinate of (0.33, 0.37), which resides in the white region of the CIE chromaticity diagram (the inset in Fig. 10A).
Fluorescence decay analysis (Fig. 10B) of the 700 °C calcined h-(Gd0.95Dy0.05)PO4 found that both the 478 and 573 emissions can be fitted with the following second-order exponential equation:50
| | I(t) = A1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−t/τ1) + A2exp(−t/τ2) exp(−t/τ1) + A2exp(−t/τ2) | (3) |
where
I(t) is the luminescence intensity at time
t,
A1 and
A2 are pre-exponential constants, and
τ1 and
τ2 stand for the decay time for the exponential components. The fitting results are summarized in the inset of
Fig. 10B together with the adjusted
R-squared values of fitting. The goodness of fit can be recognized from the
R2 values of ∼1 in each case. The results thus indicate that the fluorescence decay can be considered as a superposition of two main components in each case, with the faster one most likely arising from the surface related Dy
3+ and the slower one being related to the inner-part Dy
3+. The average fluorescence lifetime (
τ*) was calculated from the following equation to be ∼234.7 and 229.4 ns for the 478 and 573 nm emissions, respectively.
| | | τ* = (A1τ21 + A2τ22)/(A1τ1 + A2τ2) | (4) |
The slightly shorter lifetime of the 573 nm emission is due to the involvement of surface-region Dy3+ ions.
4. Conclusion
Quasi-equiaxed nanocrystals of monoclinic (m-) and hexagonal (h-) structured REPO4 (RE = La–Dy) have been successfully synthesized via ethylene glycol (EG)-assisted solvothermal reaction. It was concluded that EG effectively inhibits the 1D growth of REPO4 by selectively capping the {001} facets and meanwhile affects the nucleation density, leading to substantial size/morphology variation of the resultant nanocrystals. Owing to lanthanide contraction, the orthophosphates of La–Sm and Gd–Dy crystallize in the monoclinic and hexagonal systems, respectively. The solubility of REPO4 gradually increases from La–Dy, and as a result successively larger phosphate crystals were obtained. Calcination at 700 °C did not affect the phase purity and crystal morphology of h-GdPO4 but greatly enhanced the excitation and emission intensities of photoluminescence. The 4F9/2 → 6H13/2 (573 nm) and 4F9/2 → 6H15/2 (478 nm) emissions are similarly strong for h-(Gd0.95Dy0.05)PO4, owing to the nanosize effect, though the Dy3+ activators were assumed to have centrosymmetric D2 point symmetry. The h-(Gd0.95Dy0.05)PO4 phosphor has a CIE coordinate of (0.33, 0.37) under 274 nm excitation of the host Gd3+ ions, which falls in the white light region. The 478 and 573 nm fluorescence of h-(Gd0.95Dy0.05)PO4 both decay in a bi-exponential manner and have average lifetime values of ∼234.7 and 229.4 ns, respectively.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was supported in part by the National Natural Science Foundation of China (Grant No. 51672039, 51702020), the Fund of the State Key Laboratory of Advanced Technologies for Comprehensive Utilization of Platinum Metals (SKL-SPM-201505), and the Fundamental Research Funds for the Central Universities (Grants No. N160206001 and N160204008). Z. H. Wang acknowledges financial support from the China Scholarship Council for his overseas Ph. D. study (Contract No. 201606080030) and the assistance of H. B. Xu (National Engineering Research Center for Rare Earth Materials, Beijing, China) for his help with lifetime measurement.
References
- Y. Hikichi and T. Nomura, J. Am. Ceram. Soc., 1987, 70, 252 Search PubMed.
- F. H. Firsching and S. N. Brune, J. Chem. Eng. Data, 1991, 36, 93 CrossRef CAS.
- Y. Guo, P. Woznicki, A. Barkatt, E. E. Saad and I. G. Talmy, J. Mater. Res., 1996, 11, 639 CrossRef CAS.
- A. Dwivedi, A. K. Singh and S. B. Rai, Dalton Trans., 2014, 43, 15906 RSC.
- M. Mondal, V. K. Rai, C. Srivastava, S. Sarkar and R. Akash, J. Appl. Phys., 2016, 120, 233101 CrossRef.
- A. P. Alivisatos, Science, 1996, 271, 933 CAS.
- H. X. Mai, Y. W. Zhang, L. D. Sun and C. H. Yan, Chem. Mater., 2007, 19, 4514 CrossRef CAS.
- K. Rajesh, P. Mukundan, P. K. Pillai, V. R. Nair and K. G. K. Warrier, Chem. Mater., 2004, 16, 2700 CrossRef CAS.
- Z. H. Wang, J.-G. Li, Q. Zhu, X. D. Li and X. D. Sun, Dalton Trans., 2016, 45, 5290 RSC.
- P. C. de Sousa Filho and O. A. Serra, J. Phys. Chem. C, 2011, 115, 636 CAS.
- S. R. Liviano, F. J. Aparicio, T. C. Rojas, A. B. Hungría, L. E. Chinchilla and M. Ocaña, Cryst. Growth Des., 2012, 12, 635 Search PubMed.
- X. Wang, J. Zhuang, Q. Peng and Y. D. Li, Nature, 2005, 473, 121 CrossRef PubMed.
- W. D. Shi, S. Y. Song and H. J. Zhang, Chem. Soc. Rev., 2013, 42, 5714 RSC.
- Y. P. Fang, A. W. Xu, R. Q. Song, H. X. Zhang, L. P. You, J. C. Yu and H. Q. Liu, J. Am. Chem. Soc., 2003, 125, 16025 CrossRef CAS PubMed.
- Z. Y. Huo, C. Chen, D. R. Chu, H. H. Li and Y. D. Li, Chem. – Eur. J., 2007, 13, 7708 CrossRef CAS PubMed.
- Y. X. Ni and J. M. Hughes, Am. Mineral., 1995, 80, 21 CAS.
- W. O. Milligan and D. F. Mullica, Inorg. Chim. Acta, 1982, 60, 39 CrossRef CAS.
- Y. W. Zhang, Z. G. Yan, L. P. You, R. Si and C. H. Yan, Eur. J. Inorg. Chem., 2003, 6, 4099 CrossRef.
- Z. H. Wang, J.-G. Li, Q. Zhu, B.-N. Kim and X. D. Sun, Mater. Des., 2017, 126, 115 CrossRef CAS.
- Y. P. Fang, A. W. Xu, A. M. Qin and R. J. Yu, Cryst. Growth Des., 2005, 5, 1221 CAS.
- Y. J. Zhang and H. M. Guan, J. Cryst. Growth, 2003, 256, 156 CrossRef CAS.
- M. Yang, H. P. You, K. Liu, Y. H. Zheng, N. Guo and H. J. Zhang, Inorg. Chem., 2010, 49, 4996 CrossRef CAS PubMed.
- Z. A. Peng and X. G. Peng, J. Am. Chem. Soc., 2001, 123, 1389 CrossRef CAS.
- Z. A. Peng and X. G. Peng, J. Am. Chem. Soc., 2002, 124, 3343 CrossRef CAS PubMed.
- Y. D. Yin and A. P. Alivisatos, Nature, 2005, 739, 664 CrossRef PubMed.
- S. L. Gai, C. X. Li, P. P. Yang and J. Lin, Chem. Rev., 2004, 114, 2343 CrossRef PubMed.
- X. H. Yin, Q. Zhao, B. Q. Shao, W. Lv, Y. H. Li and H. P. You, CrystEngComm, 2014, 16, 5543 RSC.
- B. Q. Shao, Q. Zhao, N. Guo, Y. C. Jia, W. Z. Lv, M. M. Jiao, W. Lv and H. P. You, CrystEngComm, 2013, 15, 5776 RSC.
- A. I. Becerro, S. R. Liviano, A. J. F. Carrión and M. Ocaña, Cryst. Growth Des., 2013, 13, 526 CAS.
- A. Bao, H. Yang, C. Y. Tao, Y. Zhang and L. L. Han, J. Lumin., 2008, 128, 60 CrossRef CAS.
- N. K. Sahu, R. S. Ningthoujam and D. Bahadur, J. Appl. Phys., 2012, 112, 014306 CrossRef.
-
R. L. Rich, Inorganic Reactions in Water, Heidelberg, Berlin, Germany, 2007 Search PubMed.
- Y. Yang, Q. C. Zhang, Y. Y. Pan, L. S. Long and L. S. Zheng, Chem. Commun., 2015, 51, 7317 RSC.
- C. J. Jia, L. D. Sun, L. P. You, X. C. Jiang, F. Luo, Y. C. Pang and C.-H. Yan, J. Phys. Chem. B, 2005, 109, 3284 CrossRef CAS PubMed.
- M. Yang, H. P. You, Y. C. Jia, H. Qiao, N. Guo and Y. H. Song, CrystEngComm, 2011, 13, 4046 RSC.
- H.-I. Chen and H.-Y. Chang, Colloids Surf., A, 2004, 242, 61 CrossRef CAS.
- J.-G. Li, Q. Zhu, X. D. Li, X. D. Sun and Y. Sakka, Acta Mater., 2011, 59, 3688 CrossRef CAS.
-
J. W. Mullin, Crystallization, Woburn, 4th edn, 2001 Search PubMed.
- A. Hezel and S. D. Ross, Spectrochim. Acta, 1966, 22, 1949 CrossRef CAS.
-
J. A. Gadsden, Infrared Spectra of Minerals and Related Inorganic Compounds, Butterworth, Newton, 1975 Search PubMed.
- R. Kijkowska, E. Cholewka and B. Duszak, J. Mater. Sci., 2003, 38, 223 CrossRef CAS.
- J. Fujita, A. E. Martell and K. Nakmoto, J. Chem. Phys., 1962, 36, 339 CrossRef CAS.
- R. H. Hauge, J. W. Kauffman and J. L. Margrave, J. Am. Chem. Soc., 1980, 102, 6005 CrossRef CAS.
- X. F. Shi, J.-G. Li, Q. Zhu, X. D. Li and X. D. Sun, J. Alloys Compd., 2017, 695, 1984 CrossRef CAS.
- J. Yang, C. X. Li, Z. W. Quan, C. M. Zhang, P. P. Yang, Y. Y. Li, C. C. Yu and J. Lin, J. Phys. Chem. C, 2008, 112, 12777 CAS.
- Y. C. Li, Y. H. Chang, Y. F. Lin, Y. S. Chang and Y. J. Lin, J. Alloys Compd., 2007, 439, 367 CrossRef CAS.
- B. R. Judd, Phys. Rev., 1962, 127, 750 CrossRef CAS.
- G. S. Ofelt, J. Chem. Phys., 1962, 37, 511 CrossRef CAS.
- J. K. Li, J.-G. Li, S. H. Liu, X. D. Li, X. D. Sun and Y. Sakka, J. Mater. Chem. C, 2013, 1, 7614 RSC.
- Q. F. Guo, Q. D. Wang, L. W. Jiang, L. B. Liao, H. K. Liu and L. F. Mei, Phys. Chem. Chem. Phys., 2016, 18, 15545 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ce01880k |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *d,
Qi
Zhu
ab,
Byung-Nam
Kim
c,
Xudong
Sun
abe and
Ji-Guang
Li
*d,
Qi
Zhu
ab,
Byung-Nam
Kim
c,
Xudong
Sun
abe and
Ji-Guang
Li