
Selective MeCN/EtCN sorption and preferential inclusion of substituted benzenes in a cage structure with arylsulfonamide-armed anthraquinones†
Takashi
Takeda
 *a,
Shin-ichiro
Noro
b,
Takayoshi
Nakamura
b,
Yasutaka
Suzuki
c,
Jun
Kawamata
c and
Tomoyuki
Akutagawa
*a
*a,
Shin-ichiro
Noro
b,
Takayoshi
Nakamura
b,
Yasutaka
Suzuki
c,
Jun
Kawamata
c and
Tomoyuki
Akutagawa
*a
aInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, Sendai, Miyagi 980-8577, Japan. E-mail: takeda@tagen.tohoku.ac.jp
bResearch Institute of Electronic Science, Hokkaido University, Sapporo, Hokkaido 060-0820, Japan
cGraduate School of Medicine, Yamaguchi University, Yamaguchi, Yamaguchi 753-8512, Japan
First published on 2nd November 2017
Abstract
We report a series of crystal structures of arylsulfonamide-armed anthraquinones (AQs) (1–4). The arylsulfonamide-armed AQs formed orthogonal aromatic arrangements between the AQ unit and terminal aryl units due to well-defined intramolecular hydrogen bonding between the carbonyl units of AQs and the amino groups of sulfonamide units. Three disubstituted AQs 1–3 formed fundamental dimer structures, which were stabilized by intermolecular π–π interaction between AQs. Subtle differences in the dimer structures led to different packing structures. Among them, the 1,8-bis(arylsulfonamide) derivative (1) formed solvated crystals of 1·(MeCN), which exhibited reversible and selective MeCN and/or EtCN adsorption–desorption behavior. Tetra(arylsulfonamide) AQ (4) with four bulky substituents on its periphery formed various host–guest molecular crystals of 4·X2 (X = toluene, xylene, trimethylbenzenes, 1,2,3,5-tetramethylbenzene, anisole, and benzonitrile) with a rectangular zero-dimensional cage surrounded by the π-planes of 4.
Introduction
Over the past few decades, crystals with a well-defined supramolecular structure/assembly such as supramolecular cages/capsules,1 metal organic frameworks (MOFs)2 and covalent organic frameworks (COFs)3 have been vigorously studied in terms of not only basic understanding of the self-assembly process but also their application to adsorption, inclusion and separation. Control of the molecular arrangement in such supramolecular crystals has been achieved with strong interactions such as coordination bonds,4 dynamic covalent bonds5 and hydrogen bonds.6 Supramolecular capsules have been prepared by connection of the components through these bonds. Coordination bonds and dynamic covalent bonds are essential for the construction of MOFs and COFs, respectively. On the other hand, the precise arrangement of organic molecules without strong noncovalent interactions7 is difficult, and there have been few studies on their function. While covalent bonding (>300 kJ mol−1), coordination bonding (100–300 kJ mol−1)4 and electrostatic hydrogen bonding (∼10–40 kJ mol−1)6 interactions are relatively strong and directly affect crystal structures, other weak intermolecular interactions7 such as π–π stacking,8 charge-transfer (CT),9 van der Waals10 and C–H⋯π interactions11 are considered to be weak (less than 10 kJ mol−1) and only influence the closest-packing structure within crystals. The construction of unique molecular arrangements governed by weak noncovalent interactions could offer new functional crystalline materials with unique functions.Very recently, we reported the electrochemical and photophysical properties of arylsulfonamide-armed anthraquinones (AQs) 1–4.12 Noncovalent intramolecular hydrogen bonding affected the electron-accepting properties of 1–4 (Fig. 1). Preferential molecular arrangement with intramolecular hydrogen bonding between the quinone and amine units was achieved by steric-restriction of the rotation of amine units with bulky arylsulfone units, which was also demonstrated by an X-ray single-crystal structure analysis of 2. Based on the molecular geometry of 2, we expected that arylsulfonamide-armed anthraquinone derivatives would be good synthons for the construction of well-defined supramolecular assemblies governed by weak noncovalent interactions for the following reasons: (1) strong hydrogen bonding interactions are negated in an intramolecular fashion, and thus weak noncovalent interactions should govern crystal packing; (2) the rigid π frameworks of anthraquinone and terminal aryl units are arranged orthogonally via a sulfonamide spacer; (3) the electronically active anthraquinone unit and polar sulfone unit should significantly contribute to intermolecular connection.
 | ||
| Fig. 1 Molecular structures of 1–4. Ar = 4-tolyl for 1 and 2; 4-tert-butylphenyl for 3 and 4. | ||
In this work, we report the crystal structures of 1–4 and their functions. Subtle differences in the nature of the substitution lead to diverse crystal structures. Thanks to the rigid, orthogonal molecular framework of arylsulfonamide-armed AQs 1–4, weak noncovalent interactions such as π stacking, dipole–dipole interaction and intermolecular C–H⋯O interaction efficiently contribute to the determination of the crystal structures of 1–4. Significant dimer structures were formed in disubstituted AQs, which amplified the differences in the molecular structures to induce different crystal arrangements in 1–3. Among them, crystal 1 showed reversible solvent-selective adsorption and crystal 4 showed selective inclusion of substituted benzene derivatives.
Experimental section
General methods
Compounds 1–4 were prepared as described in the literature.12 Single crystals were prepared by recrystallization from the solvents summarized in Tables 1 and 2. Commercially available solvents were used as received. 1H (400 MHz) NMR spectra were recorded on a Bruker Avance III 400 NMR spectrometer. Thermogravimetric analysis was performed on a Rigaku Thermoplus EVO2 at a scan rate of 10 K min−1.| 1·(MeCN) | 1·(EtCN) | 2 | 3 | |
|---|---|---|---|---|
| Recryst. solvent | MeCN | EtCN | Toluene | MeCN |
| Chemical formula | C30H25N3O6S2 | C31H27N3O6S2 | C28H22N2O6S2 | C34H34N2O6S2 |
| Formula weight | 587.66 | 601.69 | 546.61 | 630.77 |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (#2) (#2) |
P (#2) |
P (#2) |
P21/c (#14) |
| a, Å | 8.70878(16) | 8.62524(16) | 8.13783(15) | 11.6922(2) |
| b, Å | 12.5376(2) | 12.5088(2) | 11.6634(2) | 20.8466(4) |
| c, Å | 12.6766(2) | 13.0025(2) | 13.4980(3) | 13.0536(2) |
| α, ° | 86.7619(9) | 87.7507(11) | 93.4739(9) | |
| β, ° | 79.0870(8) | 79.5340(10) | 96.7310(10) | 101.5286(7) |
| γ, ° | 86.3097(11) | 85.0219(11) | 103.8825(11) | |
| V, Å3 | 1354.89(4) | 1373.92(4) | 1229.92(4) | 3117.55(10) |
| Z | 2 | 2 | 2 | 4 |
| D calc, g cm−1 | 1.441 | 1.454 | 1.476 | 1.344 |
| μ, cm−1 | 22.14 | 21.97 | 23.81 | 19.49 |
| Refls. meas. | 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 710 710 |
15837 |
14160 |
34920 |
| Indep. refls. | 4849 | 4941 | 4398 | 5692 |
| Refls. used | 4849 | 4941 | 4398 | 5692 |
| R 1 (all) | 0.0661 | 0.0683 | 0.1037 | 0.0501 |
| wR2 (all) | 0.1506 | 0.1574 | 0.2066 | 0.1125 |
| R 1 (>2σ) | 0.0514 | 0.0521 | 0.0703 | 0.0439 |
| wR2 (>2σ) | 0.1429 | 0.1435 | 0.1761 | 0.1091 |
| GOF | 1.104 | 1.072 | 1.043 | 1.062 |
| CCDC no. | 1550096 | 1550093 | 1550105 | 1550102 |
| 4·(Toluene)2 | 4·(Anisole)2 | 4·(Fluorobenzene)2 | 4·(Benzonitrile)2 | 4·(Xylene)2 | |
|---|---|---|---|---|---|
| Recryst. solvent | Toluene | Anisole | Fluorobenzene | Benzonitrile | Xylene |
| Chemical formula | C68H74N4O10S4 | C68H76N4O12S4 | C66H70F2N4O10S4 | C68H70N6O10S4 | C70H82N4O10S4 |
| Formula weight | 1235.59 | 1269.61 | 1245.54 | 1259.58 | 1267.68 |
| Space group |
P (#2) |
P (#2) |
P (#2) |
P (#2) |
P (#2) |
| a, Å | 10.9957(8) | 10.9459(2) | 10.9315(2) | 10.9203(2) | 11.2092(4) |
| b, Å | 12.0694(11) | 12.1089(2) | 12.0707(2) | 11.9997(2) | 12.1810(4) |
| c, Å | 12.8047(7) | 12.9588(3) | 12.7651(2) | 12.8965(2) | 12.8326(4) |
| α, ° | 85.373(6) | 84.1094(13) | 84.8346(7) | 84.6483(10) | 85.5050(17) |
| β, ° | 73.757(5) | 72.9530(13) | 72.9469(7) | 73.4045(9) | 74.8198(18) |
| γ, ° | 75.131(5) | 74.0952(13) | 75.4678(7) | 75.8535(10) | 73.1335(18) |
| V, Å3 | 1576.8(2) | 1578.80(5) | 1558.54(5) | 1569.95(5) | 1618.26(9) |
| Z | 1 | 1 | 1 | 1 | 1 |
| D calc, g cm−1 | 1.301 | 1.335 | 1.327 | 1.332 | 1.301 |
| μ, cm−1 | 18.89 | 19.25 | 19.63 | 19.19 | 18.52 |
| Refls. meas. | 18025 |
18093 |
18016 |
18017 |
18885 |
| Indep. refls. | 5647 | 5678 | 5596 | 5643 | 5820 |
| Refls. used | 5647 | 5678 | 5596 | 5643 | 5820 |
| R 1 (all) | 0.0926 | 0.0696 | 0.0599 | 0.0499 | 0.1313 |
| wR2 (all) | 0.1973 | 0.1753 | 0.1341 | 0.1254 | 0.3241 |
| R 1 (>2σ) | 0.0601 | 0.0568 | 0.0473 | 0.0419 | 0.0966 |
| wR2 (>2σ) | 0.1540 | 0.1685 | 0.1262 | 0.1131 | 0.2700 |
| GOF | 1.011 | 1.140 | 1.094 | 1.098 | 1.097 |
| CCDC no. | 1550147 | 1550148 | 1550149 | 1576397 | 1576399 |
| 4·(1,2,3-Trimethylbenzene)2 | 4·(1,2,4-Trimethylbenzene)2 | 4·(pentamethylbenzene)2·(CHCl3) | 4·(Hexamethylbenzene)2 | ||
|---|---|---|---|---|---|
| Recryst. solvent | 1,2,3-Trimethylbenzene | 1,2,4-Trimethylbenzene | CHCl3/EtOH | CHCl3/EtOH | |
| Chemical formula | C72H84N4O10S4 | C72H84N4O10S4 | C77H93Cl3N4O10S4 | C78H96N4O10S4 | |
| Formula weight | 1293.72 | 1293.72 | 1469.2 | 1377.88 | |
| Space group |
P (#2) |
P (#2) |
P (#2) |
P21/n (#14) | |
| a, Å | 11.4757(5) | 11.0418(5) | 14.1225(3) | 14.0762(3) | |
| b, Å | 12.2489(5) | 12.4363(5) | 17.0279(3) | 11.0705(2) | |
| c, Å | 12.7795(6) | 12.5998(5) | 17.4930(3) | 23.2945(5) | |
| α, ° | 85.3743(19) | 88.953(2) | 81.2059(11) | ||
| β, ° | 76.1090(18) | 77.1337(19) | 69.2791(11) | 92.1146(10) | |
| γ, ° | 70.8493(19) | 77.137(2) | 72.4461(10) | ||
| V, Å3 | 1647.30(13) | 1643.53(11) | 3746.31(13) | 3627.51(13) | |
| Z | 1 | 1 | 2 | 2 | |
| D calc, g cm−1 | 1.304 | 1.308 | 1.302 | 1.261 | |
| μ, cm−1 | 18.30 | 18.34 | 26.33 | 16.92 | |
| Refls. meas. | 19124 |
18879 |
42313 |
38367 |
|
| Indep. refls. | 5918 | 5879 | 13423 |
6571 | |
| Refls. used | 5918 | 5879 | 13423 |
6571 | |
| R 1 (all) | 0.2328 | 0.1382 | 0.1338 | 0.1221 | |
| wR2 (all) | 0.4139 | 0.2879 | 0.2007 | 0.2324 | |
| R 1 (>2σ) | 0.1230 | 0.0848 | 0.0818 | 0.0870 | |
| wR2 (>2σ) | 0.3184 | 0.2278 | 0.1783 | 0.2155 | |
| GOF | 1.062 | 1.102 | 0.825 | 0.994 | |
| CCDC no. | 1576401 | 1576402 | 1576404 | 1550107 |
X-ray structural analysis
Crystallographic data for single crystals summarized in Tables 1 and 2 were collected using a diffractometer equipped with a rotating anode fitted with a multilayer confocal optic using Cu-Kα (λ = 1.54187 Å) radiation. Structure refinements were carried out using the full-matrix least-squares method on F2. Calculations were performed using the Crystal Structure and SHELX software packages.13Adsorption–desorption isotherm experiment
The adsorption isotherms for MeCN (298 K), EtCN (298 K), MeOH (288 K), and EtOH (298 K) were measured using an automatic volumetric adsorption apparatus (BELSORP-aqua, BEL Japan, Inc.). The N2 and CO2 adsorption/desorption isotherms were measured with a BELSORP-max automatic volumetric adsorption apparatus (BEL Japan) at 77 and 195 K, respectively. Before the measurements, the crystals were maintained at 373 K under a pressure of less than 10−2 Pa for 17 h to remove adsorbed molecules.Results and discussion
Packing structure
Fig. S1† shows the ORTEP drawings of bis(arylsulfonamide) AQs 1–3 and tetra(arylsulfonamide) AQ 4. Due to effective hydrogen bonding between the N–H protons and carbonyl units of anthraquinone, the effective dipole moments of sulfonamide units, and the steric repulsion between anthraquinone and terminal aryl groups, these molecules possessed a well-defined molecular geometry. Sulfone units and terminal aryl groups were oriented orthogonally with respect to the central anthraquinone π core. Arylsulfone units were oriented in opposite directions from one another so that the dipole moment could be negated intramolecularly in 1,4-disubstituted 2 and 1,5-disubstituted 3 in the respective nonsolvated crystals while both arylsulfone units were oriented in the same direction in 1,8-disubstituted 1 in the MeCN-solvated crystal (vide infra). Since the conformation of the arylsulfonamide groups of 1–4 was fixed by intramolecular N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bonding interactions, their packing structures in the crystals were mainly dominated by van der Waals interactions. Single-crystal X-ray structural analyses of crystals 1·(MeCN), 2, 3, and 4·(toluene)2 revealed the structural flexibility and diversity of the packing structures: the packing structures of molecules 1–4 were entirely different from each other. The bis(arylsulfonamide) derivatives of 1–3 formed a fundamental π-dimer structural unit (Fig. S2†), which was further connected by weak intermolecular C–H⋯OS interactions. A difference in the substituted positions of the arylsulfonamide groups affected the dimer structure, which induced a significant difference in the packing arrangement between regioisomers 1–3. The detailed packing arrangements of 2 and 3 are discussed in the ESI.†
C hydrogen bonding interactions, their packing structures in the crystals were mainly dominated by van der Waals interactions. Single-crystal X-ray structural analyses of crystals 1·(MeCN), 2, 3, and 4·(toluene)2 revealed the structural flexibility and diversity of the packing structures: the packing structures of molecules 1–4 were entirely different from each other. The bis(arylsulfonamide) derivatives of 1–3 formed a fundamental π-dimer structural unit (Fig. S2†), which was further connected by weak intermolecular C–H⋯OS interactions. A difference in the substituted positions of the arylsulfonamide groups affected the dimer structure, which induced a significant difference in the packing arrangement between regioisomers 1–3. The detailed packing arrangements of 2 and 3 are discussed in the ESI.†
Packing structure of 1·(MeCN) and its reversible MeCN/EtCN adsorption–desorption
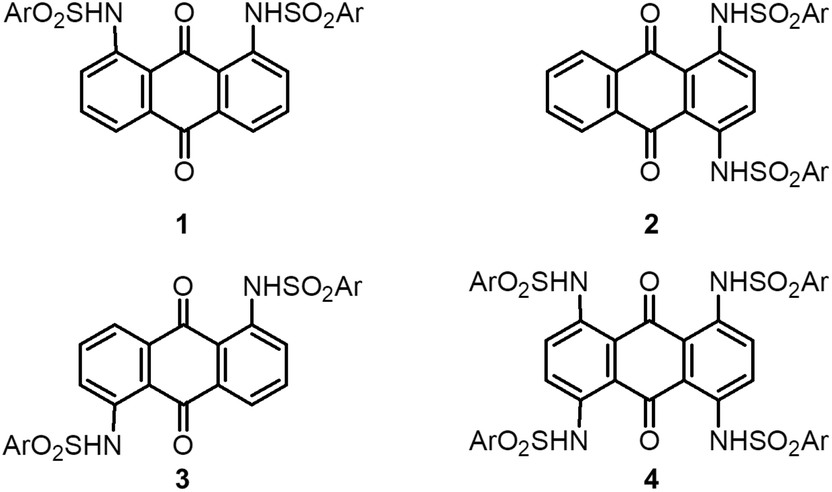
The permanent dipole moment of the π-dimer unit also affected the packing structures of molecules 1–3. Recrystallization of 1 from MeCN formed a solvated crystal of 1·(MeCN), where MeCN molecules occupied the one-dimensional (1D) channel (Fig. 2a and b) between the slipped stack of π-dimers along the vertical axis of the AQ plane. Although the molecular permanent dipole moments of 2 and 3 cancelled each other, that of 1 remained (11.1 D at the B3LYP/6-31G level with the molecular geometry determined by X-ray crystal analysis) and contributed significantly to the inclusion of MeCN within the crystal (Fig. 2c). The solvated MeCN molecules were stabilized by C–H⋯OS and CAr–H⋯NC interactions in the 1D channel. Although single crystals of 1·(EtCN) showed a packing structure similar to that of 1·(MeCN) (Fig. S3†), solvated single crystals of 1 did not form in other polar solvents, including PrCN, BuCN, PentCN, MeOH, and EtOH. This selectivity for polar solvents could be due to the compatibility of the molecular size and shape with the 1D channel.
 | ||
| Fig. 2 Crystal structure of 1·(MeCN). (a) Unit cell viewed along the a-axis. MeCN molecules are shown in a CPK model. (b) CPK drawing of the packing structure and the 1D channel along the a-axis in the absence of MeCN molecules. (c) Dipole relaxation between polar molecule 1 and MeCN within the crystal. | ||
Crystals 1·(MeCN) showed reversible and selective molecular adsorption–desorption behavior. The thermogravimetry (TG) curve of 1·(MeCN) revealed an approximately 7% weight loss at around 160 °C (Fig. S4†), which corresponds to the quantitative desorption of MeCN from the 1·(MeCN) crystals. The possible adsorption behavior of the desolvated crystal 1 was systematically examined in a variety of solvents, such as alkyl nitriles, alkyl halides, an alcohol, water, and an alkane, with different polarities, dipole moments, and sizes (Table 3). Desolvated crystals 1 were prepared by the thermal annealing of crystals 1·(MeCN) at 160 °C for 10 minutes under ambient conditions, and then exposed to vapor of the solvents in separate closet-vials. After the samples were exposed to the vapor overnight, the solvent readsorption behavior was confirmed by TG measurements. The desolvated crystals 1 show reversible and quantitative adsorption behavior only for MeCN and EtCN, to give 1·(MeCN) and 1·(EtCN), respectively. Other solvents did not show such behavior. Large alkyl nitriles such as PrCN, BuCN, and PentCN, as well as EtOH, PrCl, PrBr, PrI, and H2O, also did not show adsorption behavior.
To the best of our knowledge, such MeCN- and EtCN-selective adsorption molecular crystalline materials have not yet been reported. While the adsorption of several alkyl nitriles with coordination polymers has been reported, including Cu(II)–1,2-bis(4-pyridyl)ethane,14 Zn(II)–(5-azido isophthalic acid)[1,4-bis(4-pyridyl)-2,3-diaza-1,3-butadiene],15 and Mg(II)–bis(1,4-benzenedicarboxylate)(4,4′-dipyridyl-N,N′-dioxide),16 these materials also adsorb other organic solvents14 or long-chain nitrile derivatives.15,16
To evaluate the crystallinity and selectivity of adsorption behavior, powder X-ray diffraction (PXRD) was performed and solvent adsorption–desorption isotherms of MeCN and EtCN were examined for crystals 1. Fig. 3a summarizes the PXRD patterns of crystals 1·(MeCN), desolvated crystals 1 after thermal annealing, and the readsorbed sample of 1·(MeCN) and the simulated pattern of crystals 1·(MeCN) based on the single-crystal X-ray structural analysis. The thermal annealing of crystals 1·(MeCN) at 433 K revealed a shift and disappearance of Bragg diffraction peaks, which was consistent with a change in the packing structure from 1·(MeCN) to 1 while maintaining high crystallinity. After readsorption of the MeCN molecule, the PXRD pattern was completely consistent with that of as-grown 1·(MeCN), suggesting a reversible structural transformation between desolvated crystals 1 and solvated 1·(MeCN) upon the adsorption–desorption of MeCN. Fig. 3b shows the adsorption–desorption isotherms of desolvated crystals 1 for MeCN and EtCN along with those for MeOH and EtOH for comparison. The ratio of adsorption–desorption molecules (nads) to host molecule 1 was plotted with respect to a relative pressure P/P0. Gate-opening MeCN adsorption behavior appeared at P/P0 = ∼0.7 and the amount of saturated adsorption was similar to 1.0 mol mol−1. Once a MeCN molecule was adsorbed into crystal 1, the adsorption state was stably maintained until P/P0 ∼ 0.04 in the desorption process. A similar gate-opening adsorption behavior for EtCN was also observed at P/P0 ∼ 0.5 with an increase in nads, which was lower than the gate-opening pressure of MeCN. This gate-opening sorption mechanism with huge hysteresis for adsorption–desorption was characteristic of adsorption accompanied by a structural change. Both of the guest molecules MeCN and EtCN interacted by multiple weak van der Waals interactions within the 1D channel, as demonstrated by the single-crystal X-ray analysis of 1·(MeCN), and the structural transformation of flexible van der Waals crystals was a driving force to achieve gate-opening 1D channels. The origin of the selective adsorption of MeCN and EtCN could be accounted for by dipole moments to open the gate and the period length of the fundamental π-dimer. While nitriles with a greater dipole moment (3.92 D for MeCN)17 could open the gate, less polar solvents, such as alcohols (1.70 D for MeOH),17 could not interact with the host molecular framework efficiently enough to open the one-dimensional channel. DFT calculation supported the affirmative MeCN/EtCN adsorption of 1 (see the ESI† for details). The period cycle (6.75 Å) of the permanent dipole moment of the fundamental π-dimer makes it possible to sort alkyl nitriles. An alkyl nitrile longer than the period cycle length could not be accommodated in the dimer and only the shorter MeCN (3.0 Å) and EtCN (4.5 Å) could be included in the dimer to open the gate.
 | ||
| Fig. 3 MeCN and EtCN adsorption–desorption behavior of desolvated crystals 1. (a) PXRD patterns of MeCN-solvated crystals before and after thermal annealing and readsorption of the crystals. (b) Adsorption–desorption isotherms of desolvated crystals 1 for MeCN, EtCN, MeOH and EtOH. | ||
Host–guest complexes of tetrasubstituted AQ 4
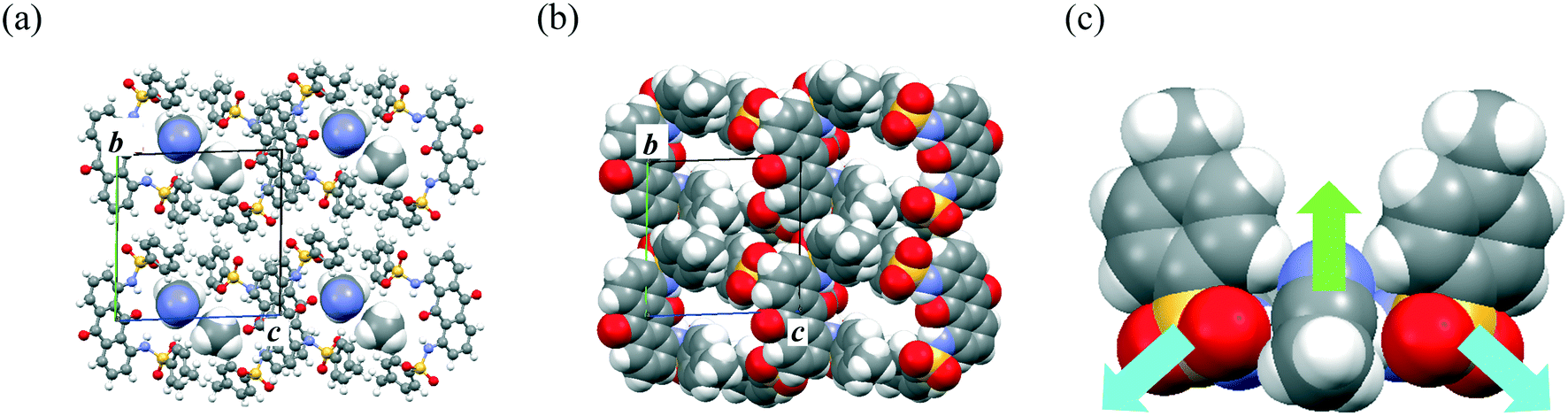
In contrast to disubstituted AQs 1–3, tetrasubstituted AQ 4 did not form a dimer structure, and instead formed a host–guest complex with substituted benzenes. Fig. 4 shows the packing structure of 4·(toluene)2. Toluene molecules formed the π-dimer, which was further stacked between the π-planes of two AQ units and surrounded by the terminal arylsulfone pillars (Form A). The toluene π-dimer was surrounded by a rectangular cage of two molecules of 4, which was elongated along the a-axis to form a 1D column. In addition to formation of the π-dimer, π–π interaction between AQ and toluene and C–H⋯OS interaction were observed. Although the toluene molecules reflected static disorder, the crystal stability was quite high without desorption of the solvated molecules from the crystals under ambient conditions. Notably, the crystals of 4 showed the preferential formation of host–guest molecular complexes with substituted-benzene derivatives in the crystalline cage. For example, not only toluene but also anisole and xylene, with electron-donating substituent(s), and fluorobenzene and benzonitrile, with electron-deficient substituents, were also included in the pillar space with the same packing arrangement (Fig. S5 and S6†).
 | ||
| Fig. 4 Host–guest molecular complexes of 4·(toluene)2 in the crystal (Form A). (a) Packing arrangement of a host–guest complex viewed along the a-axis. (b) Molecular arrangement of the toluene π-dimer within the cage encapsulated by two molecules of 4. The included toluene molecules are shown in a CPK model for clarity. | ||
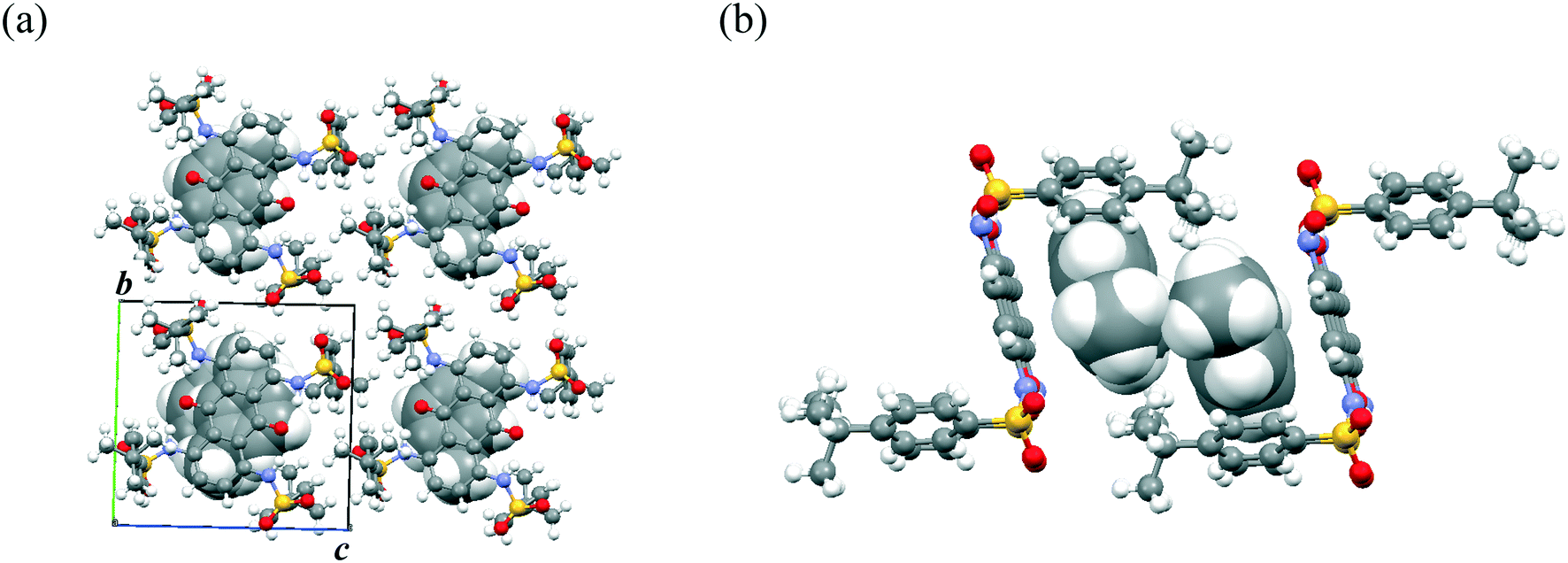
This solvent inclusion behavior is affected by the size of the benzene ring (Fig. 5a). When nonsubstituted benzene was used for recrystallization, unstable single crystals were obtained, the quality of which soon turned poor after removal from the solvent. Thus, a too small guest molecule destabilized the crystals of this host–guest complex. In contrast, flexible distortion of the crystalline cage enabled the formation of the same host–guest complexes even with bulky benzene derivatives including trimethylbenzenes and 1,2,3,5-tetramethylbenzene (Fig. S7†). The host rectangular cage unit transmuted to modulate the void space for guest molecules (368–470 Å for each cage) so that more bulky benzene derivatives could be accommodated. When the bulky benzenes were included, two modes of stretching were possible for this supramolecular cage: (a) stretching along the long axis consisting of sulfone units and tert-butylphenyl groups [observed in 4·(xylene)2 and 4·(1,2,3-trimethylbenzene)2] or (b) slanting of the rectangular cage [observed in 4·(1,2,4-trimethylbenzene)2]. Much more bulky guest molecules including durene, pentamethylbenzene, and hexamethylbenzene further modulated the guest inclusion cage itself to another type (Form B in Fig. 5b). The bulky benzene guest is stabilized by π–π interaction and also van der Waals interaction between tert-butyl groups of host molecule 4 in Form B. The balance of the size of the guest and van der Waals interactions between the host and the guest play important roles in determining the packing arrangement of 4·(arene)2 in the crystal. In contrast to Form A, the packing arrangement of host molecule 4 in Form B does not have enough flexibility to modulate the void space. Under recrystallization from durene and pentamethylbenzene, the corresponding solvated crystals were obtained, where the solvate molecules filled the spare void space.
 | ||
| Fig. 5 (a) Relationship between host–guest crystals of 4·(arene)2. (b) Packing arrangement of 4·(hexamethylbenzene) viewed along the b-axis. Hexamethylbenzene molecules are shown in a CPK model. | ||
We next examined selective inclusion of the solvent into the pillar space in crystal 4. We chose to examine methylbenzene derivatives because the selective inclusion of liquid molecules with a similar size and polarity is very challenging. Fig. 6 shows the 1H NMR spectrum of crystal 4 obtained by recrystallization from equal volumes of a solvent mixture of toluene, mesitylene, 1,2,3-trimethylbenzene, and 1,2,4-trimethylbenzene. As a result, significant preferential inclusion of 1,2,3-trimethylbenzene was observed. The ratio of guest molecules to host 4 decreased in the order of 1,2,3-trimethylbenzene (100%), 1,2,4-trimethylbenzene (33%), toluene (22%), and mesitylene (10%). Highly polarized 1,2,3-trimethylbenzene (0.62 D at the B3LYP/6-31G level with the optimized structure) was preferentially included in the cage of 4, suggesting that dipole–dipole interaction within the crystalline cage space plays an important role.
 | ||
| Fig. 6 1H NMR spectrum of crystals 4 obtained by recrystallization from a mixed solvent in CDCl3. The peaks of protons for guest molecules were assigned by measurements of 1H NMR spectra of the respective solvents in CDCl3. Numbers in the magnified spectra correspond to the respective trimethylbenzenes. | ||
Conclusion
In summary, arylsulfonamide-substituted AQ derivatives 1–4 were investigated in terms of their crystal structures with selective and reversible MeCN/EtCN adsorption–desorption behavior and selective host–guest complex formation with benzene derivatives. The crystal structures of these arylsulfonamide-armed AQs showed diverse packing structures with 1D channels and 0D cages. 1,8-Bis(arylsulfonamide) derivative 1 formed solvated crystals of 1·(MeCN), and exhibited reversible and selective gate-opening MeCN and/or EtCN adsorption–desorption behavior while maintaining high crystallinity. Both the dipole moment and size of guest molecules were essential for achieving selective adsorption–desorption behavior. Tetra(arylsulfonamide) derivative 4 formed various host–guest molecular crystals of 4·X2 (X = toluene, xylene, trimethylbenzenes, 1,2,3,5-tetramethylbenzene, anisole, fluorobenzene and benzonitrile) within a rectangular 0D cage surrounded by the π-planes of 4. This pillar-shaped void space consisting of two molecules of 4 preferentially accommodates more polar 1,2,3-tetramethylbenzene compared to other methylbenzene derivatives due to dipole–dipole interaction of the guest molecules in the pillar.Designed soft organic crystals with adsorption/inclusion properties based on the control of weak noncovalent interactions should increase the scope of potential functional crystalline materials from rigid crystalline materials such as MOFs and COFs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Number JP17K05769.References
- (a) S. Zarra, D. M. Wood, D. A. Roberts and J. R. Nitschke, Chem. Soc. Rev., 2015, 44, 419–432 RSC; (b) D. Ajami, L. Liu and J. Rebek Jr., Chem. Soc. Rev., 2015, 44, 490–499 RSC; (c) F. Hof, S. L. Craig, C. Nuckolls and J. Rebek Jr., Angew. Chem., Int. Ed., 2002, 41, 1488–1508 CrossRef CAS; (d) T. Mitra, K. E. Jelfs, M. Schmidtmann, A. Ahmed, S. Y. Chong, D. J. Adams and A. I. Cooper, Nat. Chem., 2013, 5, 276–281 CrossRef CAS PubMed; (e) T. Heinz, D. M. Rudkevich and J. Rebek Jr., Nature, 1998, 394, 764–766 CrossRef CAS.
- Review: J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- Reviews: (a) S. Das, P. Heasman, T. Ben and S. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef CAS PubMed; (b) S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC; (c) N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS.
- (a) J. A. Martinho Simões and J. L. Beauchamp, Chem. Rev., 1990, 90, 629–688 CrossRef; (b) W. Partenheimer, Inorg. Chem., 1972, 11, 743–746 CrossRef CAS; (c) W. Partenheimer and E. F. Hoy, Inorg. Chem., 1973, 12, 2805–2809 CrossRef CAS.
- Review: (a) S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef; (b) Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC; (c) A. Wilson, G. Gasparini and S. Matile, Chem. Soc. Rev., 2014, 43, 1948–1962 RSC.
- (a) P. A. Kollman and L. C. Allen, Chem. Rev., 1972, 72, 283–303 CrossRef CAS; (b) G. A. Jeffery, An Introduction to Hydrogen Bonding, Oxford University Press, New York, 1997 Search PubMed.
- Reviews: (a) P. A. Kollman, Acc. Chem. Res., 1977, 10, 365–371 CrossRef CAS; (b) K. Muller-Pethlefs and P. Hobza, Chem. Rev., 2000, 100, 143–167 CrossRef; (c) J. N. Israelachvilli, Intermolecular and Surface Forces 3rd Ed., Elsevier, 2011 Search PubMed.
- (a) S. K. Burley and G. A. Petsko, Science, 1985, 229, 23–28 CAS; (b) C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS.
- (a) L. J. Andrews, Chem. Rev., 1954, 54, 713–776 CrossRef CAS; (b) G. Briegleb, Angew. Chem., Int. Ed. Engl., 1964, 3, 617–632 CrossRef; (c) C. K. Prout and J. D. Wright, Angew. Chem., Int. Ed. Engl., 1968, 7, 659–667 CrossRef CAS; (d) H. A. Bent, Chem. Rev., 1968, 68, 587–648 CrossRef CAS.
- C. J. Van Oss, M. K. Chaudhury and R. J. Good, Chem. Rev., 1988, 88, 927–941 CrossRef CAS.
- (a) M. Nishio, M. Hirota and Y. Umezawa, The C–H/π Interaction: Evidence, Nature, and Consequences, Wiley-VCH, 1998 Search PubMed; (b) M. Nishio, CrystEngComm, 2004, 6, 130–158 RSC.
- T. Takeda, Y. Suzuki, J. Kawamata, S. Noro, T. Nakamura and T. Akutagawa, Phys. Chem. Chem. Phys., 2017, 19, 23905–23909 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- S. Noro, T. Akutagawa and T. Nakamura, Chem. Commun., 2010, 46, 3134–3136 RSC.
- S. Mistry, R. Hota and S. Natarajan, Chem. – Asian J., 2017, 12, 1807–1815 CrossRef CAS PubMed.
- J. Xu, Y. Yu, G. Li, S. Wang, Y. Liu, D. Liu and C. Wang, RSC Adv., 2016, 6, 104451–104455 RSC.
- C. P. Smith, Dielectric Behavior and Structure, McGraw-Hill, 1955 Search PubMed; Tables of Electric Dipole Moments, ed. L. G. Wesson, The Technology Press, MIT, 1948 Search PubMed; A. L. McCellan, Table of Experimental Dipole Moments, W. H. Freeman, San Francisco, 1963 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ORTEP drawings of 1–4. Dimer structures and packing arrangements of 2 and 3. Crystal structure of 1·(EtCN). Thermogravimetry curve of 1·(MeCN) crystals. DFT energy calculation of 1·(MeCN) and 1·(EtCN). Packing arrangements of crystals 4·(xylene)2, 4·(anisole)2, 4·(fluorobenzene)2, 4·(benzonitrile)2 and 4·(trimethylbenzene)2. Gas adsorption experiment with crystals 1 and 3. CCDC 1550093, 1550096, 1550102, 1550105, 1550107, 1550147–1550149, 1576397, 1576399, 1576401, 1576402 and 1576404. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ce01752a |
| This journal is © The Royal Society of Chemistry 2018 |