*
Corresponding authors
a
Department of Applied Chemistry, Graduate School of Engineering, Tokyo University of Agriculture and Technology, 2-24-16 Naka-cho, Koganei, Tokyo 184-8588, Japan
E-mail:
muraoka@go.tuat.ac.jp
b
Frontier Research Institute for Interdisciplinary Sciences, Tohoku University, 6-3 Aramaki-Aza-Aoba, Aoba-ku, Sendai 980-8578, Japan
E-mail:
okmasaki@mail.tagen.tohoku.ac.jp
c
Department of Chemistry, School of Science, Tokai University, Kitakaname, Hiratsuka-shi, Kanagawa 259-1292, Japan
d
Graduate School of Science and Engineering, Kinki University, 3-4-1 Kowakae, Higashi-Osaka, Osaka 577-8502, Japan
e
Institute of Multidisciplinary Research for Advanced Materials, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai 980-8577, Japan
f
Institute of Global Innovation Research, Tokyo University of Agriculture and Technology, Japan


![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X)NH2 (X = O or NH) groups is effective in promoting oxidative protein folding. In particular, a thiol compound coupled with a guanidyl (X = NH) group significantly accelerates the rates of folding processes and enhances the yields of native proteins.
X)NH2 (X = O or NH) groups is effective in promoting oxidative protein folding. In particular, a thiol compound coupled with a guanidyl (X = NH) group significantly accelerates the rates of folding processes and enhances the yields of native proteins.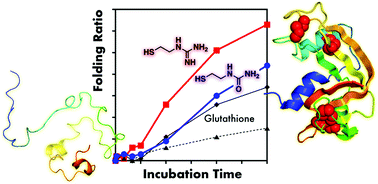
 Please wait while we load your content...
Please wait while we load your content...

