
Mechanochemical carbon–carbon bond formation that proceeds via a cocrystal intermediate†
Krunoslav
Užarević  *a
and
Ivan
Halasz
*a
*a
and
Ivan
Halasz
*a
*a
and
Ivan
Halasz
*a
Abstract
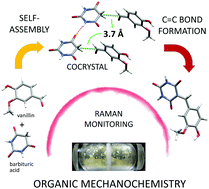
We report the first cocrystal as an intermediate in a solid-state organic reaction wherein molecules of barbituric acid and vanillin assume a favorable orientation for the subsequent Knoevenagel condensation.
 Please wait while we load your content...
Please wait while we load your content...

