
MIL-100Cr with open Cr sites for a record N2O capture†

Abstract
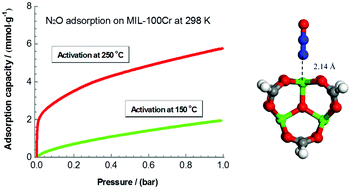
Nitrous oxide (N2O) is considered as the third most important greenhouse gas after carbon dioxide and methane and needs to be removed from air. Herein, we reported the metal–organic framework MIL-100Cr with open Cr sites for record N2O capture capacities of 5.78 mmol g−1 at 298 K and 8.25 mmol g−1 at 273 K, respectively. DFT calculations showed that the static binding energy of N2O on the open-Cr site is notably higher than that of N2, 72.5 kJ mol−1vs. 51.6 kJ mol−1, which enforces MIL-100Cr to exhibit extremely high N2O/N2 ideal adsorbed solution theory (IAST) gas separation selectivity up to 1000.
 Please wait while we load your content...
Please wait while we load your content...

