
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTaming a silyldiium cation and its reactivity towards sodium phosphaethynolate†
André
Hermannsdorfer
a,
Douglas W.
Stephan
 b and
Matthias
Driess
*a
b and
Matthias
Driess
*a
aInstitut für Chemie, Technische Universität Berlin, Strasse des 17. Juni 135, 10623 Berlin, Germany. E-mail: matthias.driess@tu-berlin.de
bDepartment of Chemistry, University of Toronto, 80 St. George Street, Toronto, Ontario M5S 3H6, Canada
First published on 9th November 2018
Abstract
A dicationic bis(NHC)-stabilised silyldiium complex, [bis(NHC)-SiPh2]2+ (72+) (bis(NHC) = [CH2(NC3H2NDipp)2], Dipp = 2,6-iPr2C6H3), was synthesised for the first time. It reacts with sodium phosphaethynolate (NaOCP) as a source of monoanionic phosphorus to give the P-insertion product [bis(NHC)-PSiPh2]+ (8+). The latter comprises a seven-membered heterocycle containing a Si–P moiety which can easily be desilylated when exposed to dichlorophosphanes as exemplified by the synthesis of the diphosphanide cations [bis(NHC)-PPCy]+ (9+) and [bis(NHC)-PPPh]+ (10+).
Since the first synthesis of a stable imidazol-2-ylidene in 1991,1 N-heterocyclic carbenes (NHCs) have been employed for the stabilisation of various main group elements in unusual electronic states,2 among them silicon and phosphorus.3 Commonly, the ligand is introduced in its deprotonated form, as “free” NHC, often followed by reduction reactions. However, in some cases, the use of a “free” NHC leads to side reactions, like for example the reduction of PIII centres to PI, and is therefore not applicable.4,5 Other strategies have been developed to overcome this limit, such as adduct formation with silyl moieties and subsequent desilylation of the NHC. This approach has been used, inter alia, by Weigand et al. to access imidazoliumyl substituted phosphines by reacting the NHC-silyl adducts 1[OTf] and 2[OTf] with PCl3 liberating Me3SiCl (Fig. 1).5,6 The same group has also described the imidazoliumyl-functionalised diphosphorus cations 3+ and 6+, which are depicted in Fig. 1.7,8 A related synthetic approach to obtain carbene–phosphinidyne transition metal complexes was used by Tamm and co-workers who introduced the IPr–PSiMe3 adduct (IPr = 1,3-bis-(Dipp)imidazol-2-ylidene, Dipp = 2,6-iPr2C6H3) as a silylated ligand precursor.9 In this case, the silicon moiety is also introduced as a leaving group and removed in the course of the synthetic route. Accordingly, the solvolysis of IPr–PSiMe3 afforded IPr–PH, which had been prepared originally by transferring parent phosphinidene (:PH) from a silylene to IPr.10 The same species can also be obtained by reacting sodium phosphaethynolate (NaOCP) with the imidazolium salt [IPr–H][Cl].11 Compared to other synthetic routes, the use of NaOCP as monoanionic phosphorus transfer reagent is advantageous by means of atom economy as only one equivalent of CO and the corresponding Na salt are generated in a salt metathesis reaction as by-products. By taking advantage of this approach some novel species were synthesised, among them the NHC–phosphinidene adducts IPr–PEPh3 (E = Ge, Sn).12
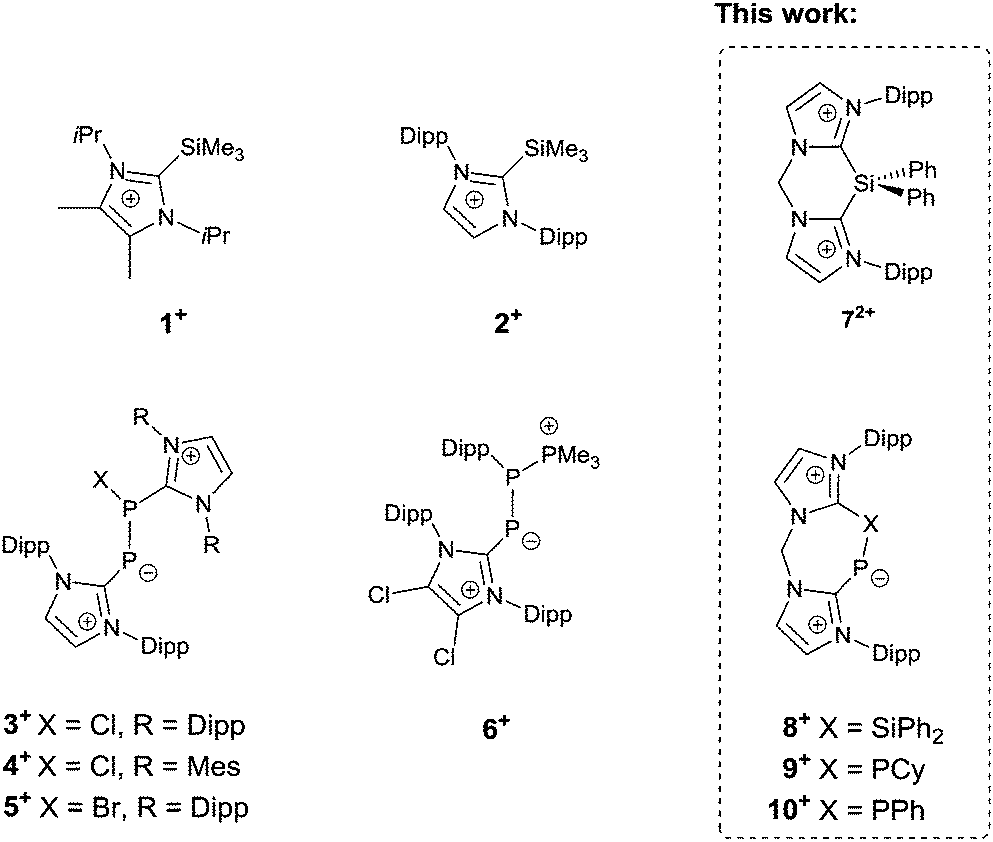 | ||
| Fig. 1 Selected known cationic imidazoliumyl-stabilised silicon and phosphorus compounds and the new compounds 72+, 8+, 9+, and 10+. | ||
Herein we report the synthesis of the novel bis(NHC)-silyldiium salt 7[OTf]2 (bis(NHC) = [CH2(NC3H2NDipp)2]) and its reactivity towards NaOCP which affords the cyclic bis(NHC)-PSiPh2 adduct 8[OTf]. The latter can be employed as a precursor for unprecedented seven-membered heterocycles by subsequent desilylation with dichlorophosphanes. This is illustrated by the reaction of 8[OTf] with RPCl2 (R = Cy, Ph), which gives rise to the phosphinidene–phosphanide cations 9+ and 10+.
The reaction of Ph2Si(OTf)2 with an equimolar amount of bis(NHC) in diethyl ether afforded the desired salt 7[OTf]2 in 84% yields (Scheme 1). The observed resonance in the 29Si NMR spectrum in CD3CN at δ = −36.7 ppm lies within the range reported for four-coordinate SiIV centres.13 Indeed, X-ray structural analysis of single crystals grown in concentrated acetonitrile solutions showed no close contact between the triflate anions and the central Si atom (Fig. 2). Moreover, the C–N bond lengths in the N–C–N moieties of the imidazoliumyl rings (average d(C–N) = 1.35 Å) express strong C–N double-bond character and thus suggest that the positive charges are delocalized over the imidazoliumyl substituents. These C–N distances are comparable to those reported for the imidazolium salt [IPr–H]Cl.14 In order to investigate the desilylation of 72+ we exposed 7[OTf]2 to PhPCl2 in o-C6H4F2 at 55 °C for three days. As expected this afforded the salt [bis(NHC)(PPh)][OTf]2 (Scheme S1, ESI†) whose synthesis we have described previously from bis(NHC) and PhPCl2.15 This desilylation complies with reported reactions of NHC-silyl adducts with halogenated phosphanes that have been used in the literature to access different carbene-stabilised phosphorus compounds,5,6 and, in this regard, the “SiPh2” moiety in 72+ only serves as a leaving group.
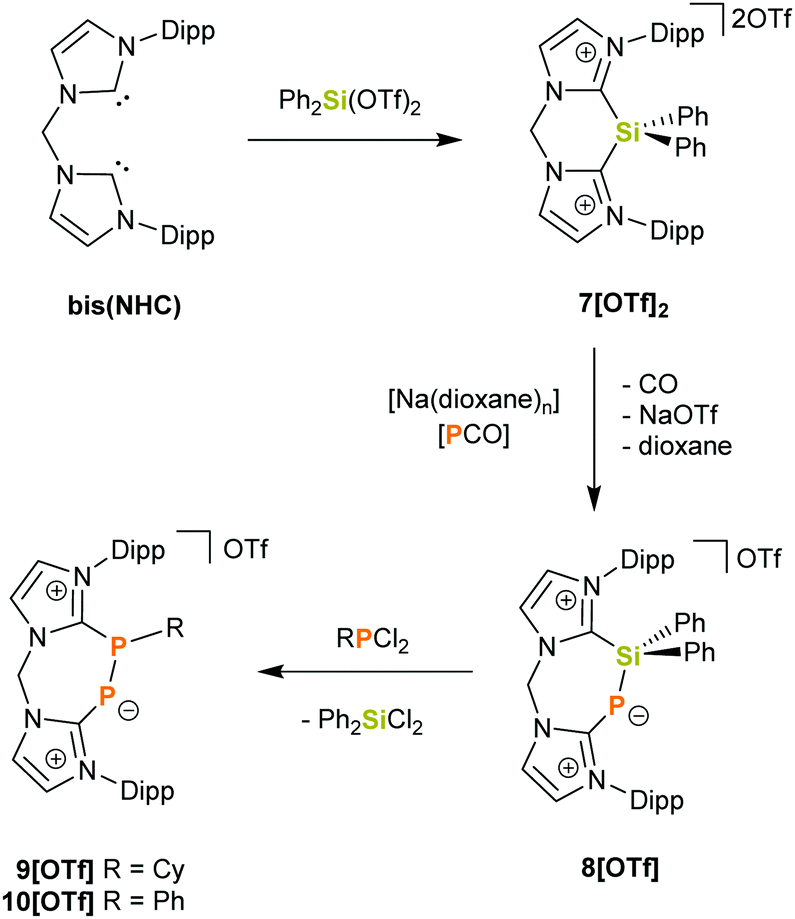 | ||
| Scheme 1 Synthetic route to compounds 7[OTf]2, 8[OTf], 9[OTf] and 10[OTf]. (Dipp = 2,6-iPr2C6H3; Cy = Cyclohexyl). | ||
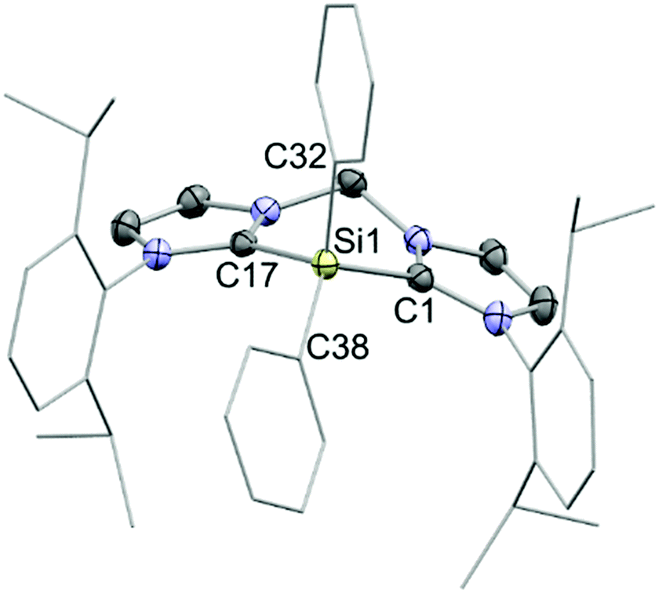 | ||
| Fig. 2 Molecular structure of the cation 72+ in 7[OTf]2·2(CH3CN), (thermal ellipsoids are given at 50% probability; H atoms are omitted for clarity); selected experimental bond lengths (Å) and angles (°); C1–Si1 1.9033(15), C17–Si1 1.8919(15), Si1–C32 1.8535(16), Si1–C38 1.8471(15), C17–Si1–C1 94.8(6), C32–Si1–C1 110.69(6), C38–Si1–C32 113.82(7). | ||
Nevertheless expecting the silicon centre to be electrophilic and thus, to allow for unprecedented synthetic pathways, we probed the reactivity of the system towards the nucleophilic phosphaethynolate anion. While literature compound IPr–PSiMe3 was synthesized using a different approach,9 IPr–PEPh3 (E = Ge, Sn),12 IPr–PH11 and an amino(phosphanylidene phosphorene)-stabilised germylene16 were obtained by using NaOCP as P− transfer reagent after CO loss at elevated temperatures. Interestingly, when mixing 7[OTf]2 with [Na(dioxane)n][PCO] (molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in CH2Cl2 at ambient temperature an immediate colour-change from ocher to red occurred, together with gas evolution (CO). Full conversion to 8[OTf] (Scheme 1) was evidenced by the rise of one singlet resonance with a characteristic high-field shift of δ = −161.0 ppm in the 31P{1H} NMR spectrum which is in the same range as for related carbene-stabilised phosphinidene adducts.9,12,17 Both 1H and 13C NMR spectra reflect the presence of two chemically inequivalent imidazoliumyl moieties and confirm that decarbonylation has occurred in the course of the reaction. Compared to the starting material 7[OTf]2, the 29Si NMR spectrum of 8[OTf] shows a low-field shifted doublet resonance at δ = −16.1 ppm (1JSiP = 85 Hz). Unlike the above-mentioned literature reactions, the formation of 8[OTf] (including CO elimination) proceeds rapidly at ambient temperature and the product could be isolated in good yields (85%). No further reaction with excess NaOCP was observed, even upon addition of up to one more molar equivalent of NaOCP.
:1) in CH2Cl2 at ambient temperature an immediate colour-change from ocher to red occurred, together with gas evolution (CO). Full conversion to 8[OTf] (Scheme 1) was evidenced by the rise of one singlet resonance with a characteristic high-field shift of δ = −161.0 ppm in the 31P{1H} NMR spectrum which is in the same range as for related carbene-stabilised phosphinidene adducts.9,12,17 Both 1H and 13C NMR spectra reflect the presence of two chemically inequivalent imidazoliumyl moieties and confirm that decarbonylation has occurred in the course of the reaction. Compared to the starting material 7[OTf]2, the 29Si NMR spectrum of 8[OTf] shows a low-field shifted doublet resonance at δ = −16.1 ppm (1JSiP = 85 Hz). Unlike the above-mentioned literature reactions, the formation of 8[OTf] (including CO elimination) proceeds rapidly at ambient temperature and the product could be isolated in good yields (85%). No further reaction with excess NaOCP was observed, even upon addition of up to one more molar equivalent of NaOCP.
Finally, the connectivity of 8+ was confirmed by a single crystal X-ray structure determination and evidences this rare example of a seven-membered ring that contains a Si–P moiety (Fig. 3). The CNHC–P bond length (1.770(2) Å) compares well to those in similar NHC-phosphinidene adducts9,12 whereas the P–Si distance (d(P–Si) = 2.1987(7) Å) in 8+ is slightly contracted compared to IPr–PSiMe3. This could be explained by the cationic charge in 8+ which makes the Si centre more electrophilic or due to ring strain. In fact, the arrangement of the atoms in a seven-membered ring leads to a distortion of the ligand backbone with respect to 72+, as visualized by comparison of the tilt angle α between the two planes defined by the imidazoliumyl rings (compare Fig. 2 and 3). While α amounts to 31.4° for 72+, α adds up to 55.3° for 8+. Furthermore, the P-insertion leads to different bonding situations in the N–C–N moieties of the two imdazoliumyl-rings. The C–N bond lengths in the Si-bonded ring (average d(C–N) = 1.35 Å) are well comparable to those in 72+, whereas the C–N distances in the P-bonded ring are somewhat elongated (average d(C–N) = 1.37 Å). This possibly reflects contributions of the canonical structures 8b+ and 8c+ to the ground state (Fig. 4).‡ The distinct 13C NMR shifts of CNHC–P (δ = 170.8 ppm, 1JCP = 102.1 Hz) and CNHC–Si (δ = 151.8 ppm) also mirror these differences.
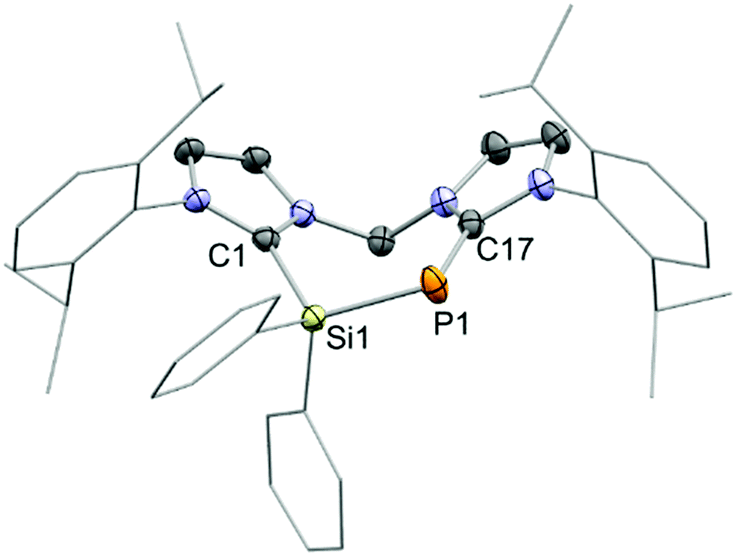 | ||
| Fig. 3 Molecular structure of the cation 8+ in 8[OTf]·(CH3CN), (thermal ellipsoids are given at 50% probability; H atoms are omitted for clarity); selected experimental bond lengths (Å) and angles (°); Si1–P1 2.1987(7), C1–Si1 1.913(2), C17–P1 1.770(2), C1–Si1–P1 111.55(7), C17–P1–Si1 105.98(7). | ||
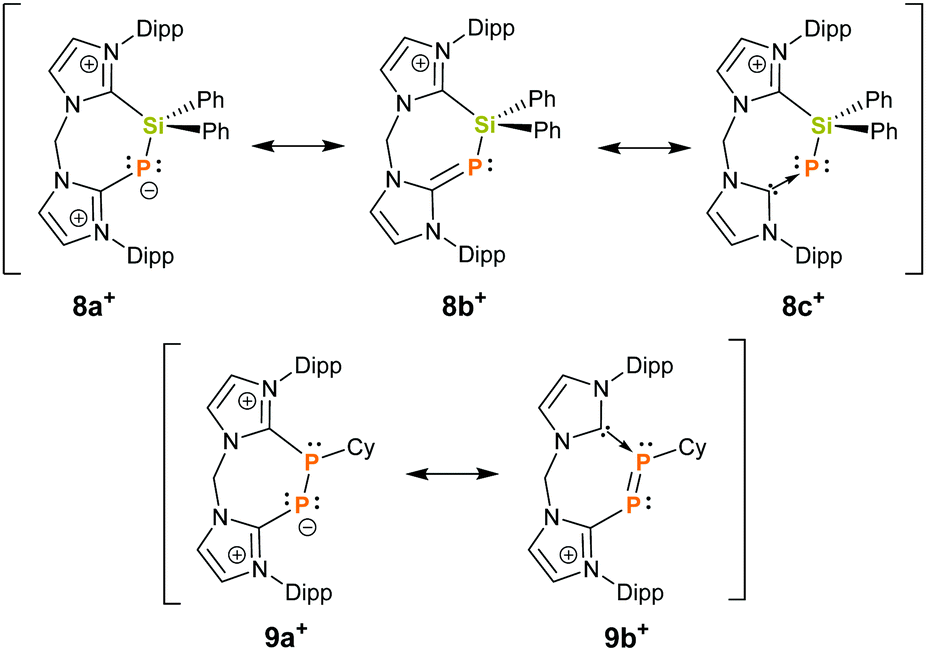 | ||
| Fig. 4 Selected resonance structures for 8+ (top) and 9+ (bottom). | ||
In order to investigate whether the reaction of 72+ with NaOCP is a particular case, we synthesised the related non-cyclic NHC-silyl cation [IPr–SiPh3]+ from an equimolar mixture of Ph3SiOTf and IPr in diethyl ether. The product precipated from the reaction mixture in form of colourless microcrystals and was characterised by heteronuclear NMR spectroscopy and high-resolution mass spectrometry indicating that the Si atom is tetracoordinate in solution. Subsequently, mixing equimolar amounts of [IPrSiPh3]OTf and NaOCP in THF led to formation of the rearrangement product IPr![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CP–O–SiPh3 (Scheme S2, ESI†) which was obtained previously by Grützmacher and co-workers from in situ generated Ph3Si-PCO and IPr.12 This finding underlines the unique properties of 72+, which are most likely due to the arrangement in a cyclic system and the presence of a second positive charge. Furthermore, compared to IPr the higher steric bulk of the bis(NHC) might influence the spatial approach of the PCO− nucleophile. Additionally, Tamm et al. reported on the dissociation equilibrium of [(NHC)SiMe3]OTf (forming NHC + Me3SiOTf) which could occur for [IPrSiPh3]OTf, as well.18 Owing to the chelating nature of the bis(NHC) ligand the respective equilibrium is most likely completely in favour of the dicationic adduct in the case of 7[OTf]2.
CP–O–SiPh3 (Scheme S2, ESI†) which was obtained previously by Grützmacher and co-workers from in situ generated Ph3Si-PCO and IPr.12 This finding underlines the unique properties of 72+, which are most likely due to the arrangement in a cyclic system and the presence of a second positive charge. Furthermore, compared to IPr the higher steric bulk of the bis(NHC) might influence the spatial approach of the PCO− nucleophile. Additionally, Tamm et al. reported on the dissociation equilibrium of [(NHC)SiMe3]OTf (forming NHC + Me3SiOTf) which could occur for [IPrSiPh3]OTf, as well.18 Owing to the chelating nature of the bis(NHC) ligand the respective equilibrium is most likely completely in favour of the dicationic adduct in the case of 7[OTf]2.
Formally, cation 8+ can be considered as a silylated chelate ligand consisting of a NHC and NHC-phosphinidene donor site. Therefore, we investigated the reaction of 8+ with dichlorophosphanes RPCl2 (R = Cy, Ph) at ambient temperature in order to elucidate whether this compound can be desilylated. Indeed, this led to fast and quantitative conversion of 8+ to 9+ (R = Cy) and 10+ (R = Ph), respectively (Scheme 1), as evidenced by the rise of two new doublet resonances in the respective 31P{1H} NMR spectra. The signals with δ = −66.5 ppm and −97.5 ppm (10+: δ = −65.0 ppm, −102.4 ppm) can be assigned to the three-coordinate P atom and to the di-coordinate phosphinidenyl moiety, respectively. These values are in excellent agreement with those reported for the related species 6+ (δ = −62.1, −108.1 ppm),8 but differ somewhat from what was observed for 4+ (δ = 138.0, −22.1 ppm)19 and 5+ (δ = 145.4, −7.6 ppm)20 which might be due to the presence of halogen atoms in the structures of 4+ and 5+ (Fig. 1). Additionally, the observed coupling constants (9+: |1JPP| = 453.5 Hz and 10+: |1JPP| = 478.8 Hz) are much larger than those accounted for comparable systems with P–P single bonds, where |1JPP| ranges between 235 Hz and 391 Hz.7,8,19–21 This could be attributed to the arrangement of the P2-unit in a rigid cyclic system, considering that 1JPP is strongly influenced by internal rotation around the P–P bond.22 In fact, the values of 1JPP come close to such that were reported for diphosphenes of the type [(NHC)P = PR]x+ (R = Aryl, NHC′ and x = 1, 2) which range between 543 Hz and 578 Hz.8,19,23 In this regard, the observed 1JPP might be an indication for partial P–P double-bond character, arising from contributions of resonance structure 9b+ to the ground state (Fig. 4). However, the 31P chemical shifts for [(NHC)P = PR]x+ were observed at substantially lower fields (δ(31P) = 398 to 452 ppm) compared to 9+ and 10+ what indicates that the double-bond character is not very pronounced.8,19,23 In the 13C NMR spectrum of 9[OTf] two doublet of doublet resonances with δ = 169.2 ppm (1JCP = 145.2 Hz, 2JCP = 10.6 Hz) and 151.5 ppm (1JCP = 76.2 Hz, 2JCP = 7.4 Hz) assignable to the N2C–carbon atoms evidence the presence of two chemically inequivalent imidazoliumyl moieties. Furthermore, a doublet resonance at δ = 45.1 ppm with a considerably smaller coupling constant (1JCP = 22.5 Hz) is observed for the cyclohexyl-carbon that is attached to one phosphorus atom.
An X-ray structure analysis confirmed the replacement of the SiPh2 fragment by the respective PR moieties in the molecular structures of 9+ and 10+ (Fig. 5 and Fig. S23, ESI†). As observed for cation 8+, the arrangement in a seven-membered ring leads to distortion of the bis(NHC) backbone. The tilt angles α between the two planes defined by the imidazoliumyl rings amount to 57.1° for 9+ and 70.4° for 10+ and are hence even larger than α in the structure of 8+ (compare Fig. 3, 5 and Fig. S23, ESI†). The P–P distances of 2.1527(10) Å (9+) and 2.1561(18) Å (10+) are very close to the one reported for the related cation 6+ (d(P–P) = 2.151(1) Å),8 but somewhat elongated in comparison to 4+ (d(P–P) = 2.1067(10) Å)19 and 5+ (d(P–P) = 2.096(2) Å)20 what may be due to the presence of phosphorus-bonded halogen atoms in the structures of 4+ and 5+. Furthermore, because of the chelating nature of the bis(NHC) ligand the torsion angles Φ defined by CNHC–P2–P1–CNHC (Φ(9+) = −52° and Φ(10+) = −46°) differ strongly from the corresponding Φ in the solid state structures of 4+ (Φ = 156°)19 and 5+ (Φ1 = 161° and Φ2 = 177°, two crystallographically independent molecules).20 Compared to the P–P distances in cationic NHC-stabilised diphosphenes where d(P–P) ranges between 2.038(1) Å and 2.083(2) Å8,19,23 the P–P bond lengths in 9+ and 10+ are elongated illustrating mostly P–P single bond character. It is worth mentioning that the desilylation of 8+ with dichlorophosphanes represents a viable procedure for a modular approach to synthesise novel cyclic compounds that assemble different main group heteroatoms in a seven-membered ring.
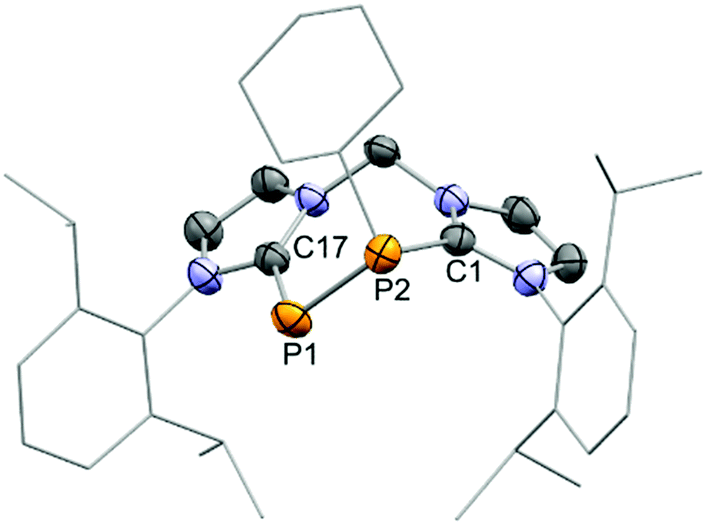 | ||
| Fig. 5 Molecular structure of the cation 9+ in 9[OTf]·(CH2Cl2)·(C7H8), (thermal ellipsoids are given at 50% probability; H atoms are omitted for clarity); selected experimental bond lengths (Å) and angles (°); P1–P2 2.1527(10), P1–P17 1.791(2), P2–C1 1.831(2), C17–P1–P2 105.42(9), C1–P2–P1 99.33(8). | ||
In summary, we have presented the synthesis of the first bis(NHC)-stabilised silyldiium salt 7[OTf]2, and shown that the particular properties of this system induce a different reactivity towards sodium phosphaethynolate than observed for the related NHC-silyl adduct [IPr–SiPh3][OTf]. The selective P-insertion reaction into one of the NHC–Si bonds in 72+ afforded cation 8+, which was subsequently desilylated with RPCl2 (R = Cy, Ph) to give the diphosphanide cations 9+ and 10+. In this way, the utility of 8[OTf] as a precursor for unprecedented seven-membered heterocyclic P–X compounds was demonstrated and is subject of further research in our laboratories.
This work was supported by the Einstein Foundation Berlin.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS.
- (a) V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed; (b) G. Frenking and N. Holzmann, Science, 2012, 336, 1394–1395 CrossRef CAS PubMed; (c) D. P. Curran, A. Solovyev, M. Makhlouf Brahmi, L. Fensterbank, M. Malacria and E. Lacôte, Angew. Chem., Int. Ed., 2011, 50, 10294–10317 CrossRef CAS PubMed; (d) H. Braunschweig, R. D. Dewhurst, K. Hammond, J. Mies, K. Radacki and A. Vargas, Science, 2012, 336, 1420–1422 CrossRef CAS PubMed; (e) Y. Xiong, S. Yao, G. Tan, S. Inoue and M. Driess, J. Am. Chem. Soc., 2013, 135, 5004–5007 CrossRef CAS PubMed; (f) M. Y. Abraham, Y. Wang, Y. Xie, P. Wei, H. F. Schaefer, P. v. R. Schleyer and G. H. Robinson, Chem. – Eur. J., 2010, 16, 432–435 CrossRef CAS PubMed; (g) C. Jones, A. Sidiropoulos, N. Holzmann, G. Frenking and A. Stasch, Chem. Commun., 2012, 48, 9855–9857 RSC.
- (a) J. D. Masuda, W. W. Schoeller, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2007, 129, 14180–14181 CrossRef CAS PubMed; (b) Y. Wang, Y. Xie, M. Y. Abraham, R. J. Gilliard, P. Wei, H. F. Schaefer, P. v. R. Schleyer and G. H. Robinson, Organometallics, 2010, 29, 4778–4780 CrossRef CAS; (c) Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2008, 130, 14970–14971 CrossRef CAS PubMed; (d) J. Arduengo Anthony III, H. V. R. Dias and J. C. Calabrese, Chem. Lett., 1997, 143–144 CrossRef; (e) R. S. Ghadwal, H. W. Roesky, S. Merkel, J. Henn and D. Stalke, Angew. Chem., Int. Ed., 2009, 48, 5683–5686 CrossRef CAS PubMed; (f) Y. Xiong, S. Yao, S. Inoue, J. D. Epping and M. Driess, Angew. Chem., Int. Ed., 2013, 52, 7147–7150 CrossRef CAS PubMed; (g) Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer, P. V. R. Schleyer and G. H. Robinson, Science, 2008, 321, 1069–1072 CrossRef CAS PubMed.
- B. D. Ellis, C. A. Dyker, A. Decken and C. L. B. Macdonald, Chem. Commun., 2005, 1965 RSC.
- J. J. Weigand, K. O. Feldmann and F. D. Henne, J. Am. Chem. Soc., 2010, 132, 16321–16323 CrossRef CAS PubMed.
- F. D. Henne, A. T. Dickschat, F. Hennersdorf, K.-O. Feldmann and J. J. Weigand, Inorg. Chem., 2015, 54, 6849–6861 CrossRef CAS PubMed.
- F. D. Henne, E. M. Schnöckelborg, K. O. Feldmann, J. Grunenberg, R. Wolf and J. J. Weigand, Organometallics, 2013, 32, 6674–6680 CrossRef CAS.
- K. Schwedtmann, M. H. Holthausen, C. H. Sala, F. Hennersdorf, R. Frohlich and J. J. Weigand, Chem. Commun., 2016, 52, 1409–1412 RSC.
- A. Doddi, D. Bockfeld, T. Bannenberg, P. G. Jones and M. Tamm, Angew. Chem., Int. Ed., 2014, 53, 13568–13572 CrossRef CAS PubMed.
- K. Hansen, T. Szilvási, B. Blom, E. Irran and M. Driess, Chem. – Eur. J., 2014, 20, 1947–1956 CrossRef CAS PubMed.
- A. M. Tondreau, Z. Benko, J. R. Harmer and H. Grützmacher, Chem. Sci., 2014, 5, 1545–1554 RSC.
- Z. Li, X. Chen, Y. Li, C. Y. Su and H. Grützmacher, Chem. Commun., 2016, 52, 11343–11346 RSC.
- Y. Takeuchi and T. Takayama, The Chemistry of Organic Silicon Compounds, John Wiley & Sons, Ltd., 1998, pp. 267–354 Search PubMed.
- A. J. Arduengo III, R. Krafczyk, R. Schmutzler, H. A. Craig, J. R. Goerlich, W. J. Marshall and M. Unverzagt, Tetrahedron, 1999, 55, 14523–14534 CrossRef.
- M. Mehta, T. C. Johnstone, J. Lam, B. Bagh, A. Hermannsdorfer, M. Driess and D. W. Stephan, Dalton Trans., 2017, 46, 14149–14157 RSC.
- N. Del Rio, A. Baceiredo, N. Saffon-Merceron, D. Hashizume, D. Lutters, T. Müller and T. Kato, Angew. Chem., Int. Ed., 2016, 55, 4753–4758 CrossRef CAS PubMed.
- M. Balmer and C. von Hänisch, Z. Anorg. Allg. Chem., 2018, 29, 4778 Search PubMed.
- M. F. Silva Valverde, E. Theuergarten, T. Bannenberg, M. Freytag, P. G. Jones and M. Tamm, Dalton Trans., 2015, 44, 9400–9408 RSC.
- A. Doddi, D. Bockfeld, M. K. Zaretzke, C. Kleeberg, T. Bannenberg and M. Tamm, Dalton Trans., 2017, 46, 15859–15864 RSC.
- J. B. Waters, T. A. Everitt, W. K. Myers and J. M. Goicoechea, Chem. Sci., 2016, 7, 6981–6987 RSC.
- (a) A. Beil, R. J. Gilliard and H. Grutzmacher, Dalton Trans., 2016, 45, 2044–2052 RSC; (b) Y. Wang, H. P. Hickox, Y. Xie, P. Wei, D. Cui, M. R. Walter, H. F. Schaefer and G. H. Robinson, Chem. Commun., 2016, 52, 5746–5748 RSC.
- (a) S. Burck, K. Götz, M. Kaupp, M. Nieger, J. Weber, J. Schmedt auf der Günne and D. Gudat, J. Am. Chem. Soc., 2009, 131, 10763–10774 CrossRef CAS PubMed; (b) S. Duangthai and G. A. Webb, Org. Magn. Reson., 1983, 21, 199–202 CrossRef CAS; (c) A. H. Cowley and W. D. White, J. Am. Chem. Soc., 1969, 91, 1917–1921 CrossRef CAS.
- O. Back, B. Donnadieu, P. Parameswaran, G. Frenking and G. Bertrand, Nat. Chem., 2010, 2, 369–373 CrossRef CAS PubMed.
- (a) G. Frenking, Angew. Chem., Int. Ed., 2014, 53, 6040–6046 CrossRef CAS PubMed; (b) D. Himmel, I. Krossing and A. Schnepf, Angew. Chem., Int. Ed., 2014, 53, 370–374 CrossRef CAS PubMed; (c) D. Himmel, I. Krossing and A. Schnepf, Angew. Chem., Int. Ed., 2014, 53, 6047–6048 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra and crystallographic details. CCDC 1866468–1866471. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc07632d |
| ‡ A comprehensive discussion on the bonding features of NHC-element adducts can be found in the literature.24 |
| This journal is © The Royal Society of Chemistry 2018 |