
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric synthesis of Rauhut–Currier-type esters via Mukaiyama–Michael reaction to acylphosphonates under bifunctional catalysis†
Víctor
Laina-Martín
 a,
Roberto
del Río-Rodríguez
a,
Sergio
Díaz-Tendero
bcd,
Jose A.
Fernández-Salas
*a and
José
Alemán
*ab
a,
Roberto
del Río-Rodríguez
a,
Sergio
Díaz-Tendero
bcd,
Jose A.
Fernández-Salas
*a and
José
Alemán
*ab
aDepartamento de Química Orgánica (Módulo 1), Facultad de Ciencias, Universidad Autónoma de Madrid, 28049-Madrid, Spain. E-mail: jose.aleman@uam.es; Web: http://www.uam.es/jose.aleman
bInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049 Madrid, Spain
cCondensed Matter Physics Center (IFIMAC), Facultad de Ciencias, Universidad Autónoma de Madrid, 28049 Madrid, Spain
dDepartamento de Química (Módulo 13), Facultad de Ciencias, Universidad Autónoma de Madrid, 28049-Madrid, Spain
First published on 23rd October 2018
Abstract
A highly enantioselective organocatalytic Mukaiyama–Michael reaction of silyloxy dienes and α,β-unsaturated acyl phosphonates under bifunctional organocatalysis is presented. The new reactivity triggered by the catalyst conducted to Rauhut–Currier type esters, via a formal conjugate addition to α,β-unsaturated esters. This protocol proceeds under mild conditions with complete regioselectivity and excellent enantiocontrol.
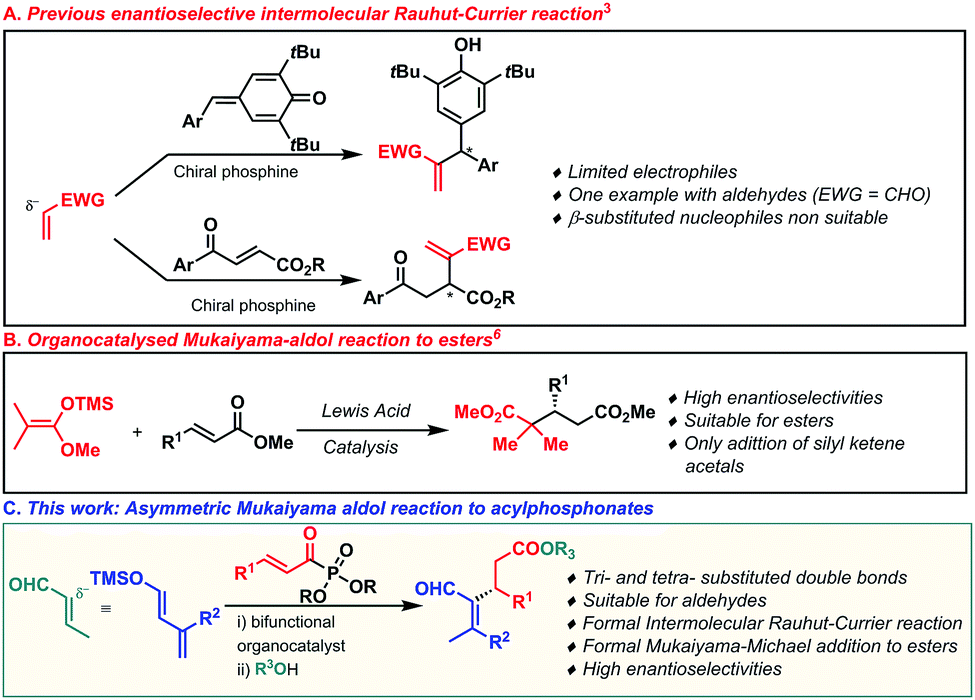
The development of efficient and practical strategies for the stereoselective construction of C–C bonds is an ongoing objective and still holds a preferred position in organic chemistry research.1 In this field, the Rauhut–Currier reaction has been widely developed over the years, becoming a powerful tool for the construction of valuable compounds from two structurally diverse olefins.2 In particular, asymmetric intramolecular Rauhut–Currier processes have been extensively explored and reported during the last few years.2a Different groups have focused their attention on developing chiral catalysts based on different nucleophilic species able to trigger the reaction.2,3 However, the intermolecular cross-asymmetric version is still underdeveloped and only a few groups have described efficient methodologies (Scheme 1, eqn (a)).3 In pursuit of this challenging objective, these research groups have described the use of well-designed multifunctional chiral phosphine-based catalysts, which have only allowed the efficient α-functionalization of 3-aroyl acrylates and para-quinone methides mainly with vinyl ketones.3b,e–h Despite these particular achievements, the enantioselective intermolecular cross Rauhut–Currier reaction has been applied successfully in the presence of certain electrophiles. Indeed, only very specific and reactive substrates such as terminal vinylic carbonyl compounds have shown sufficient reactivity. However, mono- and gem-disubstituted double bonds are more challenging electrophiles for this transformation.
 | ||
| Scheme 1 Previous works (A and B) and the present work (C). | ||
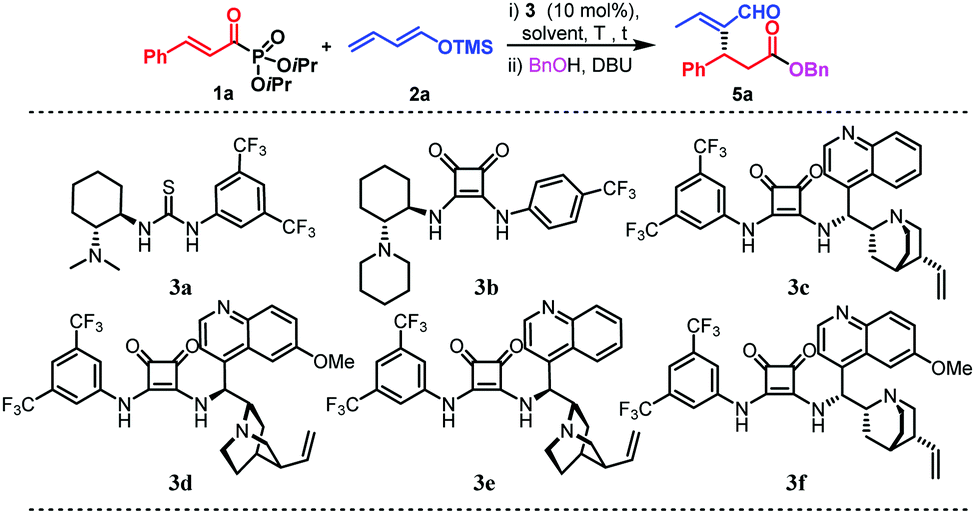
The catalytic Mukaiyama-aldol (MA) reaction has become a powerful tool for the enantioselective construction of C–C bonds.4 Despite the great efforts made in the development of MA reactions, the conjugate addition to α,β-unsaturated esters, which are difficult substrates to activate,5 still stands as an outstanding objective in asymmetric organocatalysis. Indeed, only one example has recently been reported by List and coworkers in which a silylium imidodiphosphorimidate Lewis acid has been shown to be a very efficient catalyst in the asymmetric Mukaiyama–Michael reaction to α,β-unsaturated esters (Scheme 1, eqn (b)).6 Similarly, the organocatalysed vinylogous Mukaiyama-aldol (VMA) reaction has been widely developed and has become the preferred method for the stereoselective synthesis of vinylogous aldol type products.4e,7 The orbital coefficients provoke the observed 1,5-nucleophilic attack,8 while the reactivity through the less reactive 3 position of the dienolate has been practically unexplored (Scheme 1, eqn (c)). Taking into account the challenge of expanding the asymmetric intermolecular Rauhut–Currier reaction,9 we envisioned that the addition of silyl dienol ethers to acylphosphonates,10 followed by an in situ acyl substitution, could potentially lead to Rauhut–Currier type esters. In order to achieve this objective, the nucleophilic addition to the conjugated system (1,4-addition) would need to take place through the less reactive position (1,3) of the silyl dienol ether. Herein, we describe a Mukaiyama–Michael process of silyl dienol ethers to α,β-unsaturated acyl phosphonates catalysed by a bifunctional organocatalyst.11 The special and unusual reactivity shown by these dienolates when activated by the bifunctional organocatalyst and the use of acylphosphonates as ester surrogates have allowed us to develop the formal synthesis of Rauhut–Currier-type esters, not easy to access otherwise. In addition, very challenging β-mono and β-disubstituted formal acrolein adducts have been addressed using this methodology. Based on our experience in bifunctional catalysis,9,12 we began our investigations by studying the reaction of acylphosphonate 1a as a model substrate with a trimethyl silyl dienolate derivative 2a and different thiourea and squaramide bifunctional organocatalysts (Table 1). Firstly, we examined catalysts 3a and 3b which led, after in situ treatment with BnOH and DBU, to the desired Rauhut–Currier type ester with moderate enantioselectivities (entries 1 and 2). Taking into account these preliminary results, the squaramide-quinine catalyst 3d was tested and it clearly enhanced the selectivity of the process (90% ee) (entry 3). All four squaramides bearing a member of the quinine family (3c–f) led to similar yields and enantioselectivities of the desired Rauhut–Currier ester (entries 4–7). Different solvents were then studied taking 3d as the optimised catalyst (entries 8–11). p-Xylene was considered as the optimum solvent (entry 9) as it led to higher enantioselectivity. A lower concentration and a higher catalyst loading boosted the efficiency of the reaction and yielded the desired product 5a with a 57% yield and 97% enantiomeric excess (entry 14).
|
|
|||||
|---|---|---|---|---|---|
| Entry | 3 | Solvent | t (h) | Yield 5a (%)c | eed |
| a All the reactions were performed on a 0.1 mmol scale in the presence of 3 (10 mol%) in 0.3 mL of solvent. b 3 equiv. of water was added. c Isolated yield. d Determined by SFC chromatography. e 3d (20 mol%) and 0.6 mL of solvent (0.17 M). | |||||
| 1b | 3a | THF | 36 | 36 | 59 |
| 2b | 3b | THF | 36 | 51 | 66 |
| 3b | 3d | THF | 36 | 32 | 90 |
| 4 | 3d | THF | 36 | 40 | 91 |
| 5 | 3e | THF | 36 | 34 | 89 |
| 6 | 3f | THF | 36 | 36 | −88 |
| 7 | 3c | THF | 36 | 42 | −82 |
| 8 | 3d | DCM | 36 | 36 | 94 |
| 9 | 3d | p-Xylene | 36 | 36 | 96 |
| 10 | 3d | MeCN | 36 | 35 | 90 |
| 11 | 3d | DME | 36 | 30 | 93 |
| 12e | 3d | THF | 36 | 59 | 90 |
| 13e | 3d | DCM | 48 | 30 | 93 |
| 14 | 3d | p-Xylene | 36 | 57 | 97 |
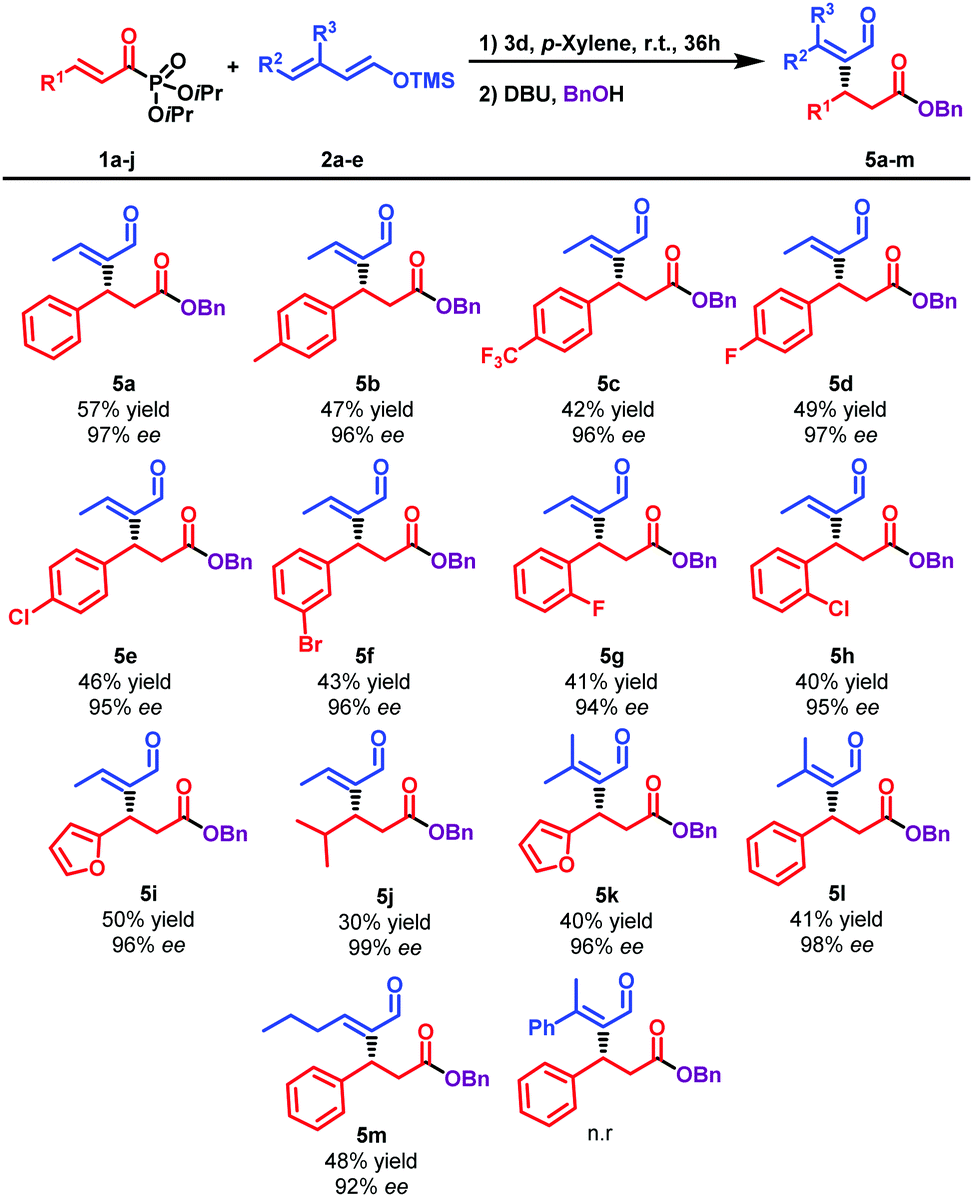
Once the reaction conditions had been optimised (entry 14, Table 1), we studied the scope of the reaction considering differently substituted α,β-unsaturated acyl phosphonates (Table 2). The Mukaiyama–Michael reaction embraced a variety of aromatics with complete regioselectivity observed in all cases. The reaction proceeded smoothly with the p-tolyl substituted double bond (5b) with an excellent enantioselectivity (96% ee). Acyl phosphonates bearing a poor aromatic ring (5c) led to the desired Rauhut–Currier product with excellent enantioselectivity, retaining the efficiency of the reaction. Differently substituted halogenated phosphonates at either the ortho, meta or the para position were very well tolerated, conducting to the β-substituted esters with excellent enantioselectivities (5d–h). The protocol enabled access to the corresponding ester in the presence of conjugated heterocyclic acylphosphonate with excellent enantioselectivities and moderate yield (5i). In the same manner, silyl dienolate reacted with an aliphatic conjugated system, affording 5j with complete enantiocontrol albeit with a slightly lower yield. Other silyl dienolates were also studied. While a silyl dienol ether bearing a longer aliphatic chain was well tolerated (5m), the presence of a phenyl group as a substituent did not conduct to the desired product. To our delight, when a 4-substituted silyloxy diene (2b) was tested, challenging β,β-disubstituted Rauhut–Currier adducts (5k–5l) were obtained with excellent enantioselectivities.
| a All the reactions were performed on a 0.1 mmol scale in 0.6 mL of p-xylene. Enantiomeric excess was determined by SFC chromatography. |
|---|
|
Interestingly, when the in situ acyl substitution was not performed under the optimised reaction conditions, the reaction conducted to the formation of the dihydropyranone derivative 4a after an in situ lactonization through intermediate I. Subsequent lactone opening in the presence of BnOH and DBU led to the desired Rauhut–Currier type ester as a unique product in quantitative yield (Scheme 2, eqn (a)). These control experiments suggested that the moderate yields obtained might come from the intrinsic instability of 4a under the reaction conditions during the long reaction times.13 Considering the whole process, which involves conjugate addition of the dienolate, cyclisation and lactone opening, the functionalised esters are obtained in synthetically useful yields. In order to explain the observed configuration, a stereochemical proposal is shown in Scheme 2. We propose that the nitrogen of the quinine moiety directly triggers the Mukaiyama–Michael reaction from the si-face of the acyl phosphonate and the silyloxy diene reacts through its carbon 3 due to the close proximity.
 | ||
| Scheme 2 (a) Control experiment and isolation of intermediate 4a. Proposed coordination model. (b) Derivatization of dihydropyranone derivatives 4 to compounds 6a and 6b. | ||
Lactone rings are structural motifs very often present in natural products. Among naturally occurring lactones, the 5,6-dihydropyran-2-one moiety is of considerable interest from a chemical and pharmacological perspectives.14 Taking advantage of the enantioselective formation of lactone 4a, 4,5-disubstituted dihydropyranones 6a and 6b were obtained with excellent enantioselectivities via simple alkene metathesis with styrene (Scheme 2, eqn (b)). The absolute configuration of the Rauhut–Currier type products series was assigned by the correlation with a known compound in the literature (6a) and by circular dichroism. It was determined as R, assuming the same stereochemical outcome for all the compounds 4, 5 and 6 (see the ESI†).
In conclusion, we have described an asymmetric Mukaiyama–Michael reaction of silyl dienolates to α,β-unsaturated acyl phosphonates as ester surrogates. The employed bifunctional organocatalyst orchestrated the approach of the silyloxy diene to the Michael acceptor, leading to the formation of Rauhut–Currier type esters. The reaction proceeded under mild conditions with complete regioselectivity, excellent enantiocontrol and a reasonably high range of α,β-unsaturated phosphonates. In addition, the methodology led to tri- and tetra-substituted Rauhut–Currier type esters.
We are grateful to the Spanish Government (CTQ2015-64561-R and CTQ2016-76061-P) and the European Research Council (ERC-CG-UNBICAT, contract number: 647550). J. A. F.-S. and V. L.-M. thank the Spanish Government for a Juan de la Cierva Contract and the Universidad Autónoma de Madrid for a predoctoral fellowship (FPI-UAM), respectively. Financial support from the Spanish Ministry of Economy and Competitiveness, through the ‘‘Maria de Maeztu’’ Program of Excellence in R&D (MDM-2014-0377), is also acknowledged. We acknowledge the generous allocation of computing time at the CCC (UAM).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) V. Farina, J. T. Reeves, C. H. Senanayake and J. J. Song, Chem. Rev., 2006, 106, 2734–2793 CrossRef CAS PubMed; (b) M. Breuer, K. Ditrich, T. Habicher, B. Hauer, M. Keßeler, R. Stürmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43, 788–824 CrossRef CAS PubMed.
- For selected general reviews on the R–C reaction, see: (a) K. Chandra Bharadwaj, RSC Adv., 2015, 5, 75923–75946 RSC; (b) P. Xie and Y. Huang, Eur. J. Org. Chem., 2013, 6213–6226 CrossRef CAS; (c) J. L. Methot and W. R. Roush, Adv. Synth. Catal., 2004, 346, 1035–1050 CrossRef CAS; (d) C. E. Aroyan, A. Dermenci and S. J. Miller, Tetrahedron, 2009, 65, 4069–4084 CrossRef CAS; (e) D. K. Nair, T. Kumar and I. N. N. Namboothiri, Synlett, 2016, 2425–2442 CAS; (f) A. M. Wensley, N. T. McDougal and S. E. Schaus, in Lewis Base Catalysis in Organic Synthesis, ed. E. Vedejs and S. E. Denmark, Wiley-VCH, 2016, pp. 655–714. For selected recent examples on the R–C reaction, see Search PubMed; (g) X. Su, W. Zhou, Y. Li and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 6874–6877 CrossRef CAS PubMed; (h) S. Takizawa, T. M.-N. Nguyen, A. Grossmann, D. Enders and H. Sasai, Angew. Chem., Int. Ed., 2012, 51, 5423–5426 CrossRef CAS PubMed.
- (a) Q. Zhao, C. Pei, X. Guan and M. Shi, Adv. Synth. Catal., 2011, 353, 1973–1979 CrossRef CAS; (b) S. Li, Y. Liu, B. Huang, T. Zhou, H. Tao, Y. Xiao, L. Liu and J. Zhang, ACS Catal., 2017, 7, 2805–2809 CrossRef CAS; (c) W. Zhou, P. Chen, M. Tao, X. Su, Q. Zhao and J. Zhang, Chem. Commun., 2016, 52, 7612–7615 RSC; (d) C. Qin, Y. Liu, Y. Yu, Y. Fu, H. Li and W. Wang, Org. Lett., 2018, 20, 1304–1307 CrossRef CAS PubMed; (e) H. Wang, K. Wang, Y. Man, X. Gao, L. Yang, Y. Ren, N. Li, B. Tang and G. Zhao, Adv. Synth. Catal., 2017, 359, 3934–3939 CrossRef CAS; (f) T.-C. Kang, L.-P. Wu, Q.-W. Yu and X.-Y. Wu, Chem. – Eur. J., 2017, 23, 6509–6513 CrossRef CAS PubMed; (g) X. Dong, L. Liang, E. Li and Y. Huang, Angew. Chem., Int. Ed., 2015, 54, 1621–1624 CrossRef CAS PubMed; (h) W. Zhou, X. Su, M. Tao, C. Zhu, Q. Zhao and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 14853–14857 CrossRef CAS PubMed.
- For selected reviews, see: (a) S. B. J. Kan, Kenneth K.-H. Ng and I. Paterson, Angew. Chem., Int. Ed., 2013, 52, 9097–9108 CrossRef CAS PubMed; (b) J.-i. Matsuo and M. Murakami, Angew. Chem., Int. Ed., 2013, 52, 9109–9118 CrossRef CAS PubMed; (c) S. Hosokawa, Tetrahedron Lett., 2018, 59, 77–88 CrossRef CAS; (d) S. V. Pansare and E. K. Paul, Chem. – Eur. J., 2011, 17, 8770–8779 CrossRef CAS PubMed; (e) M. Frías, W. Cieślik, A. Fraile, A. Rosado-Abón, A. F. Garrido-Castro, F. Yuste and J. Alemán, Chem. – Eur. J., 2018, 24, 10906–10933 CrossRef PubMed.
- For selected examples of conjugate addition of various nucleophiles to α,β-unsaturated esters, see: (a) J. Y. Kang and R. G. Carter, Org. Lett., 2012, 14, 3178–3181 CrossRef CAS PubMed; (b) Y. Kobayashi, Y. Taniguchi, N. Hayama, T. Inokuma and Y. Takemoto, Angew. Chem., Int. Ed., 2013, 52, 11114–11118 CrossRef CAS PubMed; (c) A. J. M. Farley, C. Sandford and D. J. Dixon, J. Am. Chem. Soc., 2015, 137, 15992–15995 CrossRef CAS PubMed; (d) J. Yang, A. J. M. Farley and D. J. Dixon, Chem. Sci., 2017, 8, 606–610 RSC; (e) E. Canales and E. J. Corey, J. Am. Chem. Soc., 2007, 129, 12686–12687 CrossRef CAS PubMed.
- T. Gatzenmeier, P. S. J. Kaib, J. B. Lingnau, R. Goddard and B. List, Angew. Chem., Int. Ed., 2018, 57, 2464–2468 CrossRef CAS PubMed.
- For a review, see: (a) S. V. Pansare and E. K. Paul, Chem. – Eur. J., 2011, 17, 8770–8779 CrossRef CAS PubMed; (b) M. Kalesse, M. Cordes, G. Symkenberga and H.-H. Lu, Nat. Prod. Rep., 2014, 31, 563–594 RSC; (c) S. Hosokawa, Tetrahedron Lett., 2018, 59, 77–88 CrossRef CAS . For selected examples, see: ; (d) R. P. Singh, B. M. Foxman and L. Deng, J. Am. Chem. Soc., 2010, 132, 9558–9560 CrossRef CAS PubMed; (e) N. Zhu, B.-C. Ma, Y. Zhang and W. Wang, Adv. Synth. Catal., 2010, 352, 1291–1295 CrossRef CAS; (f) V. Bhasker, M. Gravel and V. H. Rawal, Org. Lett., 2005, 7, 5657–5660 CrossRef PubMed; (g) S. E. Denmark and G. L. Beutner, J. Am. Chem. Soc., 2003, 125, 7800–7801 CrossRef CAS PubMed; (h) M. Sickert and C. Schneider, Angew. Chem., Int. Ed., 2008, 47, 3631–3634 CrossRef CAS PubMed; (i) L. Ratjen, P. García-García, F. Lay, M. E. Beck and B. List, Angew. Chem., Int. Ed., 2011, 50, 754–758 CrossRef CAS PubMed.
- S. E. Denmark, J. R. Heemstra and G. L. Beutner, Angew. Chem., Int. Ed., 2005, 44, 4682–4698 CrossRef CAS PubMed.
- M. Frías, R. Mas-Ballesté, S. Arias, C. Alvarado and J. Alemán, J. Am. Chem. Soc., 2017, 139, 672–679 CrossRef PubMed.
- Acylphosphonates have been proven to be efficient ester surrogates. For a general review, see: (a) H. Jiang, M. W. Paixao, D. Monge and K. A. Jørgensen, J. Am. Chem. Soc., 2017, 132, 2775–2783 CrossRef PubMed . For selected recent examples, see: ; (b) D. Kowalczyk and Ł. Albrecht, J. Org. Chem., 2016, 81, 6800–6807 CrossRef CAS PubMed; (c) A.-D. Steinkamp, M. Frings, I. Thomé, I. Schiffers and C. Bolm, Chem. – Eur. J., 2015, 21, 7705–7708 CrossRef CAS PubMed; (d) H.-Li Ma, L. Xie, Z. Zhang, J.-Q. Li, Z.-H. Qin and B. Fu, Adv. Synth. Catal., 2016, 358, 1011–1016 CrossRef.
- For selected reviews on bifunctional organocatalysis and hydrogen bond activation, see: (a) W.-Y. Siau and J. Wang, Catal. Sci. Technol., 2011, 1, 1298–1310 RSC; (b) P. R. Schreiner, Chem. Soc. Rev., 2003, 32, 289–296 RSC; (c) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713–5743 CrossRef CAS PubMed.
- (a) A. Parra, R. Alfaro, L. Marzo, A. Moreno-Carrasco, J. L. García Ruano and J. Alemán, Chem. Commun., 2012, 48, 9759–9761 RSC; (b) V. Marcos, J. Alemán, J. L. García Ruano, F. Marini and M. Tiecco, Org. Lett., 2011, 13, 3052–3055 CrossRef CAS PubMed; (c) F. Esteban, W. Cieślik, E. M. Arpa, A. Guerrero-Corella, S. Díaz-Tendero, J. Perles, J. A. Fernández-Salas, A. Fraile and J. Alemán, ACS Catal., 2018, 8, 1884–1890 CrossRef CAS PubMed; (d) V. Laina-Martín, J. Humbrías-Martín, J. A. Fernández-Salas and J. Alemán, Chem. Commun., 2018, 54, 2781–2784 RSC; (e) T. Rigotti, A. Casado-Sánchez, S. Cabrera, R. Pérez-Ruiz, M. Liras, V. de la Peña O'Shea and J. Alemán, ACS Catal., 2018, 8, 5928 CrossRef CAS.
- The lower yield observed for cyclic product 4a might also be due to its instability during the isolation process.
- J. A. Marco, M. Carda, J. Murga and E. Falomir, Tetrahedron, 2007, 63, 2929–2958 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc07561a |
| This journal is © The Royal Society of Chemistry 2018 |