
Intermolecular π-hole/n→π* interactions with carbon monoxide ligands in crystal structures†

Abstract
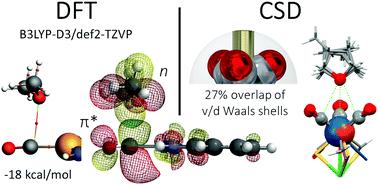
A thorough analysis of the Cambridge Structure Database reveals that intermolecular π-hole/n→π* interactions with carbon monoxide ligands are abundant in the solid state and somewhat directional, particularly with fac-like M(CO)3 fragments (P < 4.0). High level DFT calculations suggest interacting energies up to about −10 kcal mol−1 for adducts of charge neutral complexes.
 Please wait while we load your content...
Please wait while we load your content...

