
Mechanistic and asymmetric investigations of the Au-catalysed cross-coupling between aryldiazonium salts and arylboronic acids using (P,N) gold complexes†
Alexis
Tabey
,
Murielle
Berlande
,
Philippe
Hermange
 * and
Eric
Fouquet
*
* and
Eric
Fouquet
*
Univ. Bordeaux, Institut des Sciences Moléculaires, UMR-CNRS 5255, 351 Cours de la Libération, 33405 Talence Cedex, France. E-mail: philippe.hermange@u-bordeaux.fr; eric.fouquet@u-bordeaux.fr
First published on 30th October 2018
Abstract
In order to explore the different mechanisms possibly occurring in the Au-catalysed cross-coupling of ArN2BF4 and ArB(OH)2 in the presence of CsF, various stoichiometric experiments were performed on gold complexes with (P,N) ligands. Employing 2-pyridylphenyl-diphenylphosphine allowed us to suggest three different mechanistic pathways, starting either with a transmetallation step, via two consecutive single electron transfers, or by implying a transmetallation between Au(I) and Au(III) species. Moreover, when using commercially available chiral (P,N) ligands, the asymmetric formation of atropoisomeric biaryls from suitable aryldiazonium salts and arylboronic acids could be achieved with e.e. up to 26%.
The synthesis of biaryl compounds by transition metal catalysed reactions has been the subject of numerous research studies these last few decades, and various elements in the d-block were explored.1 Among them, Pd-based catalysts were undoubtedly the most successful species to enable the coupling of suitable aryl electrophiles and aryl nucleophiles with high efficiency and wide functional-group tolerance. Indeed, the palladium atom is easily able to undergo changes of its oxidation state, and its catalytic activity can be finely tuned by coordination to specifically designed monodentate/bidentate ligands.2 In contrast, the use of gold catalysts for the synthesis of aryl–aryl cross-coupling products has remained extremely challenging for a long time.3,4 Indeed, their reluctance to engage in Au(I)/Au(III) catalytic cycles has been seen as a barrier for such reactions,5 and most of the research studies in homogeneous gold catalysis were focused on employing its carbophilic Lewis acid properties for pi-activation and cascade reactions.6 However, new strategies appeared recently to overcome this issue, for example using a strong external oxidant to ensure the access to the Au(III) intermediates,7 or designing specific (P,P), (P,N) and (N,N) ligands to enable the oxidative addition onto aryl halide.8 On the other hand, employing aryldiazonium salts as electrophiles9 also led to extremely fruitful results in gold-catalysed arylation reactions. Indeed, upon thermal, light-induced, base-induced or photoredox-induced10 activation, the aryl radical generated in situ from the diazonium salt can produce the desired Au(III)-aryl species after two single electron transfers (SET).11 These complexes, obtained under extremely mild conditions from simple Au(I) precursors, are able to engage in a wide variety of transformations,12,13 including the synthesis of biaryl compounds from the cross-coupling of aryldiazonium salts (or derivatives) and arylboronic acids. Interestingly, this reaction could be catalysed by either coordinatively saturated or cationic Au species, and various conditions were reported (Scheme 1).14
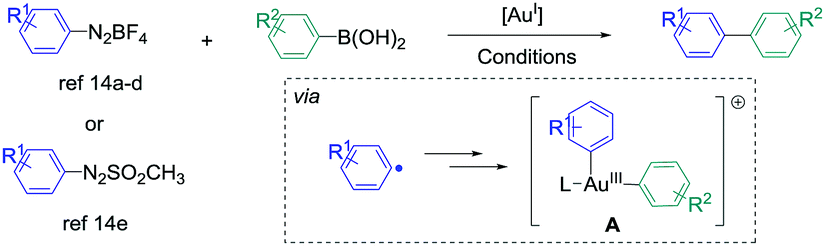 | ||
| Scheme 1 Reported gold-catalysed cross-coupling between aryldiazonium salts (or arylazosulfones) with arylboronic acids. For the exact conditions, see ref. 14. | ||
However, contrary to the well-established catalytic cycle for palladium catalysed cross-couplings involving successive oxidative addition, transmetallation and reductive elimination steps,1,2 mechanistic pathways leading to intermediate A, which undergoes the reductive elimination,15 are still unclear in these gold-catalysed aryl–aryl cross-couplings. Based on stoichiometric 1H and 31P NMR studies, transmetallation was proposed to be the first step in ref. 14a and b (catalytic cycle I). In contrast, ref. 14d and e suggested that the double SET occurred first (catalytic cycle II), taking into account the modelling experiments on related gold-catalysed transformations involving aryldiazonium salts16 and literature reports on the stoichiometric synthesis of aryl–Au(III) complexes.11 An in-between situation was described by Lee et al., whose NMR studies were in favour of either transmetallation first with PPh3AuNTf2 or oxidation first with PPh3AuCl, thus showing the complexity of mechanism determination in this reaction. Finally, despite the evident analogy with the well-known [Pd]- or [Ni]-catalysed syntheses of atropoisomeric biaryls,17 possible asymmetric induction with a gold-based system has not been described yet to our knowledge, but could open a new area of research in this field.
In view of these observations, we decided to perform stoichiometric experiments to investigate mechanistic and asymmetric possibilities of our conditions of gold-catalysed cross-coupling between aryldiazonium salts and arylboronic acids.14b Some of our preliminary experiments with PPh3AuCl as a precursor were unsuccessful,14b and all the isolated aryl–Au(III) species reported in the literature contained a supplementary ligand, i.e. a chloride ion when starting from aryldiazonium chloride,11b,e or pyridine moieties present either on the aryldiazonium tetrafluoroborate11d or on the phosphine ligand.11a,c Thus, 2-pyridylphenyl-diphenylphosphine L1 was chosen as the preferential ligand for stoichiometric studies, as the catalytic ability of L1-AuCl in the cross-coupling reaction could be confirmed (39% yield of product 3a, Scheme 2(A)). Then, transmetallation of this model gold chloride complex with phenylboronic acid 2a in the presence of two equivalents of CsF was explored following pathway I, and pleasingly it led to the desired L1AuPh (B1) with an excellent isolated yield of 94%.18 Interestingly, adding one equivalent of diazonium 1a to an acetonitrile solution of B1 under photoredox conditions produced the cross-coupling biaryl compound 3a in 30% yield (Scheme 2(B), pathway I), i.e. in a comparable yield to the one obtained employing catalytic amounts of the gold precursor (39%, Scheme 2(A)). Thus, as previously demonstrated with Ph3AuCl,14b this would suggest that Cycle I could operate in the catalytic version of this reaction. Moreover, when L1AuCl was reacted directly with 4-methoxybenzenediazonium salt 1a under blue light irradiation with Ru(bpy)3(PF6)2 as the photocatalyst, the Au(III)-complex E1 was also isolated with an excellent yield of 94% (Scheme 2(B), pathway II). Interestingly, the 31P NMR signal at 30.0 ppm was in accordance to a trans configuration between the chloro ligand and the phosphorous atom as reported previously in the literature.11b Subsequently, E1 was reacted with phenylboronic acid and cesium fluoride under the exact transmetallation conditions of pathway I, and 3a could be isolated in 35% yield in this case,19 therefore suggesting that Cycle II could also be a viable mechanism. Finally, we hypothesised that Au(I)–Au(III) ligand exchange might occur during the course of the reaction, and the reaction of L1AuPh (B1) with E1 was investigated. Interestingly, along with the homo-coupling product 4a resulting from the decomposition of E1 under the reaction conditions, the desired cross-coupling product 3a was obtained in 15% yield, confirming the possibility of a still undescribed third catalytic cycle (Cycle III). To gain further insight into the precise intermediates involved catalytically, we attempted to prepare the corresponding cationic and fluoride gold species for these three pathways, but unfortunately, it led only to unusable complex mixtures and/or degradation.
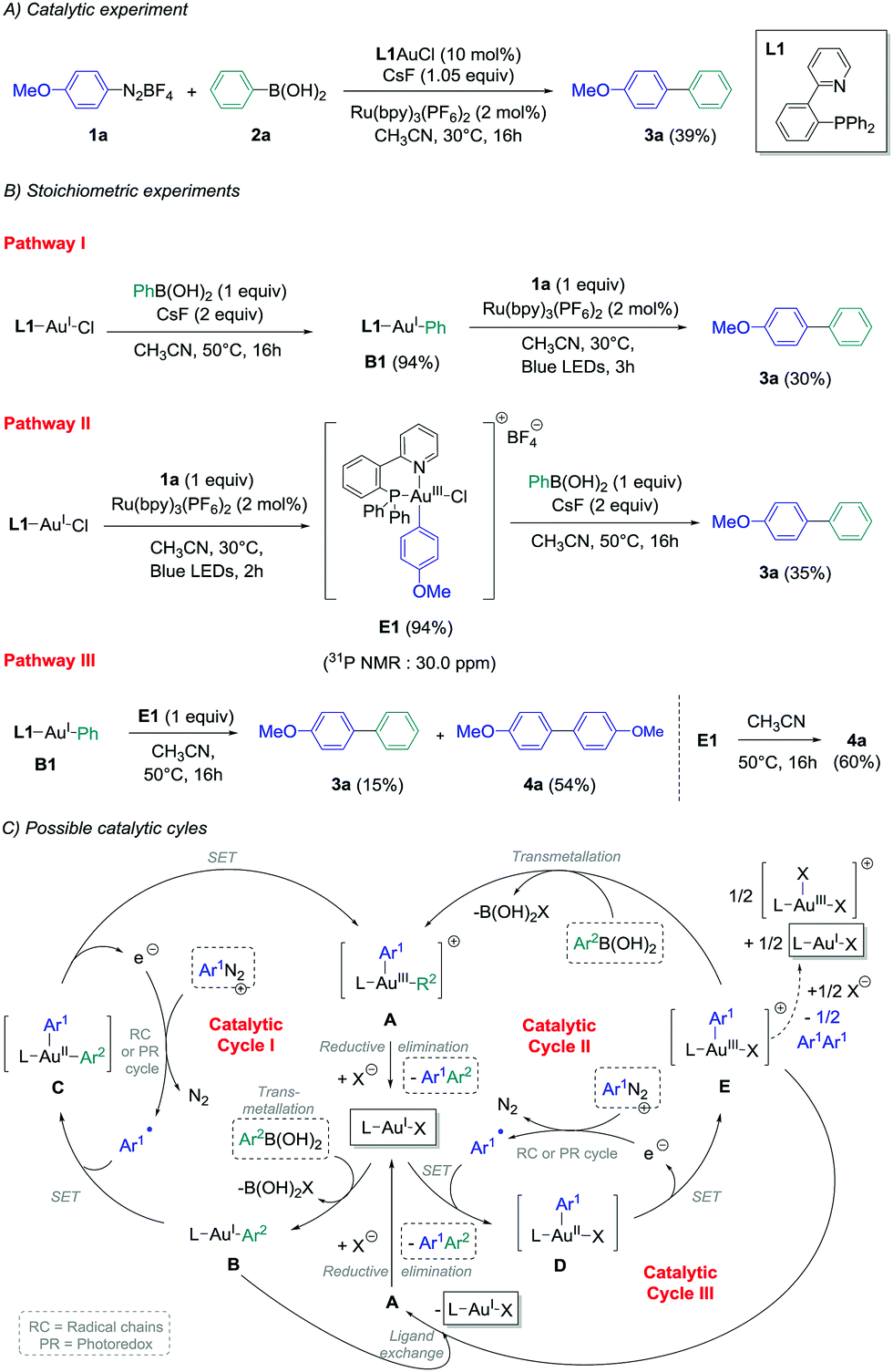 | ||
| Scheme 2 Catalytic and stoichiometric experiments of the cross-coupling between 4-methoxybenzenediazonium tetrafluoroborate 1a and phenylboronic acid 2a using L1AuCl as the gold precursor, and possible catalytic cycles. | ||
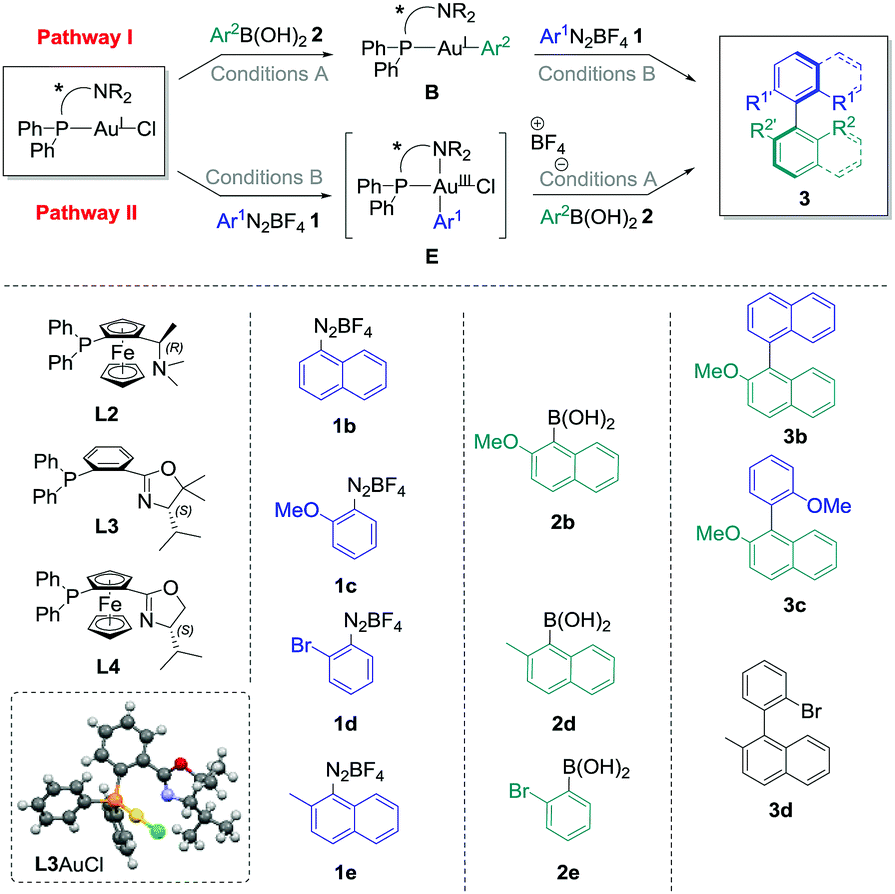
In a second part, the use of chiral catalysts was investigated in combination with more hindered starting materials to potentially produce enantioenriched atropoisomeric biaryl compounds.
The commercially available ligand (R)-(Sp)-(−)-PFNMe L2 was firstly chosen for its established ability to produce good enantioselectivities in Pd-catalysed atropoisomeric biaryl couplings.20 Pathway I was considered, and transmetallation of 2b with L2AuCl was achieved smoothly. Indeed, the Au(I) complex B2 was isolated in good crude yields (70–96%, see the ESI†) under the conditions previously developed, i.e. with two equivalents of cesium fluoride at 50 °C in acetonitrile for 16 h. Then, the reaction of this compound with one equivalent of the diazonium salt 1b under photoredox conditions provided the desired biaryl product 3b with isolated yields ranging from 28% to 41%, corresponding to a mean yield of 33% over the four experiments. Unfortunately, a very low asymmetric induction was measured for 3b using chiral HPLC (mean enantiomeric excess of +5 ± 2%, n = 4, Table 1, entry 1). On the other hand, directly adding 1b to L2AuCl in acetonitrile in the presence of 2 mol% of the Ru(bpy)32+ as a photocatalyst under blue LED irradiation leads to the major formation of the gold complex E2, characterised by its signal at 20.6 ppm in 31P NMR.‡ However, a lack of reproducibility was observed for step 2 in this case, as the desired product 3b could only be isolated for two of the six experiments, despite a full conversion of the starting complex observed in all cases (Table 1, entry 2). A mean yield of only 9% was calculated (n = 6), and a low e.e. was again measured (−8 ± 0%, n = 2), which precluded compelling mechanistic conclusions. As this poor reproducibility might be due in these conditions to the competition between pathway I, catalytic cycle III and decomposition pathways of E2, we decided to focus solely on the fully reproducible pathway I for the other experiments. (P,N) Ligands with chiral oxazoline moieties were investigated and L3AuCl was prepared,21 which produced single crystals suitable for X-ray diffraction analysis (Table 1).§ Interestingly, the crystal structure confirmed the expected absence of nitrogen coordination in this Au(I) complex. Then, the stoichiometric biaryl couplings were realised starting from this compound, and the yield of 3b obtained was similar to the one with L2AuCl for pathway I (42%, entry 3), but led to a slightly higher enantiomeric excess of 12%. Ligand L3 was selected to further investigate the substrate effects, which quickly appeared to be the determinant. Indeed, changing only the aryldiazonium salt to 2-methoxybenzenediazonium tetrafluoroborate 1c resulted in an important decrease in both the yield and enantioselectivity. For instance, the desired biaryl 3c was isolated in only 23% yield, as an almost racemic mixture (entry 4). The synthesis of 3d mediated by L3AuCl was also explored, starting either from 1d/2d or 1e/2e reagent couples (Table 1, entry 5 and entry 6, respectively). Unfortunately, all isolated yields and measured e.e. of 3d were relatively low (inferior to 10%). Finally, the best results were obtained when employing 1b, 2b and AuCl coordinated by (Sp)-1-(diphenylphosphino)-2-[(4S)-4-isopropyl-2-oxazolyl]-ferrocene L322 as starting materials, the desired biaryl product 3b being isolated in a good 44% yield over two steps, and with an improved enantiomeric excess of +26% in this case (Table 1, entry 7).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Path | L | 1 | 2 | 3 | Yielda (%) | e.e.b (%) |
| Conditions A: arylboronic acid (1–1.5 equiv.), CsF (2 equiv.), CH3CN, 50 °C, 2–72 h; conditions B: aryldiazonium tetrafluoroborate (1 equiv.), Ru(bpy)3PF6 (2 mol%), blue LED irradiation, 30 °C, 2–3 h. After step 1, the mixture was evaporated, filtered on celite, evaporated under reduced pressure and triturated in Et2O or pentane to give the crude product, which was engaged without further purification in step 2 after NMR analyses. For the exact conditions, see the ESI. Mercury drawing of one of the structure of L3AuCl obtained by X-ray diffraction analysis (50% thermal ellipsoids).a Yield of product 3 over the two steps after isolation via preparative TLC on silica gel.b Determined by chiral HPLC (measurement error of ±2%). The plus sign was arbitrarily attributed if the major enantiomer of 3 corresponded to the HLPC pic with the lower retention time, and the minus sign in the opposite case.c Mean ± standard deviation of n experiments.d Calculated for the two successful reactions out of six performed.e Product 3d was isolated in a mixture with multiple side-products, and a second purification by preparative TLC did not improved the purity of the sample.f nd: not determined. | |||||||
| 1 | I | L2 | 1b | 2b | 3b | 33 ± 6c (n = 4) | +5 ± 2c (n = 4) |
| 2 | II | L2 | 1b | 2b | 3b | 9 ± 13c (n = 6) | −8 ± 0c (n = 2d) |
| 3 | I | L3 | 1b | 2b | 3b | 42 | +12 |
| 4 | I | L3 | 1c | 2b | 3c | 23 | −3 |
| 5 | I | L3 | 1d | 2d | 3d | <10e | ndf |
| 6 | I | L3 | 1e | 2e | 3d | 5 | +3 |
| 7 | I | L4 | 1b | 2b | 3b | 44 | +26 |
To conclude, investigations on the gold-catalysed cross-coupling of aryldiazonium tetrafluoroborate salts and arylboronic acids in the presence of cesium fluoride were realised using (P,N) gold complexes. In a first part, stoichiometric experiments employing 2-pyridylphenyl-diphenylphosphine L1 as a ligand for AuCl allowed us to propose three catalytic cycles from the same precatalyst, which demonstrated a mechanistic peculiarity of gold when compared to Pd-catalysed aryl–aryl cross couplings. In a second part, preliminary stoichiometric experiments proved that Au(I) catalysts could induce the asymmetric formation of atropoisomeric biaryl compounds from suitable aryldiazonium salts and arylboronic acids with e.e. up to 26% when coordinated to chiral (P,N) structures. Due to the high substrate dependence of the outcome of the reaction, thorough research studies will be probably needed to achieve a widely applicable method. However, we believe that this work could open the way for further applications of chiral (P,N) ligands in gold-catalysed reactions implying Au(III) intermediates in their catalytic cycles.7,8,11–13,23 Such possibilities are currently being investigated by our team and will be reported in due time.
This study was supported by a public grant from the French Agence Nationale de la Recherche (AAP JCJC 2015), referenced ANR-15-CE18-0015-01 and named SuCarB. Analyses were performed at the CESAMO (UMR 5255).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) Metal-Catalyzed Cross-Coupling Reactions and More, ed. A. de Meijere, S. Bräse and M. Oestreich, Wiley-VCH, Weinheim, 2014, vol. 1–3 Search PubMed; (b) New Trends in Cross-Coupling: Theory and Applications, ed. T. J. Colacot, The Royal Society of Chemistry, Cambridge, 2015 Search PubMed.
- Palladium Reagents and Catalysts: New Perspectives for the 21st Century, ed. J. Tsuji, John Wiley & Sons Ltd, Chichester, 2004 Search PubMed.
- For an example with Au nanoparticles, see: J. Han, Y. Liu and R. Guo, J. Am. Chem. Soc., 2009, 131, 2060 CrossRef CAS PubMed.
- (a) P. Garcia, M. Malacria, C. Aubert, V. Gandon and L. Fensterbank, ChemCatChem, 2010, 2, 493 CrossRef CAS; (b) H. A. Wegner and M. Auzias, Angew. Chem., Int. Ed., 2011, 50, 8236 CrossRef CAS PubMed.
- (a) T. Lauterbach, M. Livendahl, A. Rosellón, P. Espinet and A. M. Echavarren, Org. Lett., 2010, 12, 3006 CrossRef CAS PubMed; (b) M. Livendahl, C. Goehry, F. Maseras and A. M. Echavarren, Chem. Commun., 2014, 50, 1533 RSC; (c) M. Joost, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 15022 CrossRef CAS PubMed.
- For selected references, see: (a) Modern Gold Catalyzed Synthesis, ed. A. S. K. Hashmi and F. D. Toste, Wiley-VCH, Weinheim, 2012 Search PubMed; (b) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351 CrossRef CAS PubMed; (c) N. D. Shapiro and F. D. Toste, Synlett, 2010, 675 CAS; (d) M. Rudolph and A. S. K. Hashmi, Chem. Soc. Rev., 2012, 41, 2448 RSC; (e) C. Obradors and A. M. Echavarren, Chem. Commun., 2014, 50, 16 RSC; (f) A. Fürstner, Acc. Chem. Res., 2014, 47, 925 CrossRef PubMed; (g) J. Xie, C. Pan, A. Abdukader and C. Zhu, Chem. Soc. Rev., 2014, 43, 5245 RSC; (h) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028 CrossRef CAS PubMed.
- For reviews, see: (a) M. N. Hopkinson, A. D. Gee and V. Gouverneur, Chem. – Eur. J., 2011, 17, 8248 CrossRef CAS PubMed; (b) K. M. Engle., T.-S. Mei, X. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2011, 50, 1478 CrossRef CAS PubMed ; for selected examples, see: ; (c) L. T. Ball, G. C. Lloyd-Jones and C. A. Russel, Science, 2012, 337, 1644 CrossRef CAS PubMed; (d) Q. Wu, C. Du, Y. Huang, X. Liu, Z. Long, F. Song and J. You, Chem. Sci., 2015, 6, 288 RSC; (e) A. J. Cresswell and G. C. Lloyd-Jones, Chem. – Eur. J., 2016, 22, 12641 CrossRef CAS PubMed ; for pioneering examples on related alkene–alkene couplings, see ; (f) A. S. K. Hashmi, M. C. Blanco, D. Fischer and J. W. Bats, Eur. J. Org. Chem., 2006, 1387 CrossRef CAS; (g) A. K. Hashmi, T. D. Ramamurthi and F. Rominger, J. Organomet. Chem., 2009, 592 CrossRef CAS.
- For examples, see: (a) L. Joost, A. Zeineddine, L. Estévez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 14654 CrossRef PubMed; (b) A. Zeineddine, L. Estévez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Nat. Commun., 2017, 8, 565 CrossRef PubMed; (c) N. Dwadnia, J. Roger, N. Pirio, H. Cattey, R. Ben Salem and J.-C. Hierso, Chem. – Asian J., 2017, 12, 459 CrossRef CAS PubMed; (d) M. J. Harper, C. J. Arthur, J. Crosby, E. J. Emmett, R. L. Falconer, A. J. Fensham-Smith, P. J. Gates, T. Leman, J. E. McGrady, J. F. Bower and C. A. Russell, J. Am. Chem. Soc., 2018, 140, 4440 CrossRef CAS PubMed.
- For reviews on palladium-catalysed couplings with aryldiazonium salts, see: (a) A. Roglans, A. Pla-Quintana and M. Moreno-Mañas, Chem. Rev., 2006, 106, 4622 CrossRef CAS PubMed; (b) H. Bonin, E. Fouquet and F.-X. Felpin, Adv. Synth. Catal., 2011, 353, 3063 CrossRef CAS.
- For selected recent reviews on photoredox catalysis, see: (a) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898 CrossRef CAS PubMed; (b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (c) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed.
- (a) L. Huang, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 4808 CrossRef CAS PubMed; (b) E. O. Asomoza-Solis, J. Rojas-Ocampo, R. A. Toscano and S. Porcel, Chem. Commun., 2016, 52, 7295 RSC; (c) L. Huang, F. Rominger, M. Rudolph and A. S. K. Hashmi, Chem. Commun., 2016, 52, 6435 RSC; (d) A. Tlahuext-Aca, M. N. Hopkinson, C. G. Daniliuc and F. Glorius, Chem. – Eur. J., 2016, 22, 11587 CrossRef CAS PubMed; (e) B. Dong, H. Peng, S. E. Motika and X. Shi, Chem. – Eur. J., 2017, 23, 11093 CrossRef CAS PubMed.
- For reviews, see: (a) M. N. Hopkinson, A. Tlahuext-Aca and F. Glorius, Acc. Chem. Res., 2016, 49, 2261 CrossRef CAS PubMed; (b) T. McCallum, S. Rohe and L. Barriault, Synlett, 2016, 289 Search PubMed; (c) M. Parasram and V. Gevorgyan, Chem. Soc. Rev., 2017, 46, 6227 RSC; (d) M. Zhang, C. Zhu and L.-W. Ye, Synthesis, 2017, 1150 Search PubMed; (e) M. O. Akram, S. Banerjee, S. S. Saswade, V. Bedi and N. T. Patil, Chem. Commun., 2018, 54, 11069 RSC.
- For seminal examples, see: (a) B. Sahoo, M. N. Hopkinson and F. Glorius, J. Am. Chem. Soc., 2013, 135, 5505 CrossRef CAS PubMed; (b) M. N. Hopkinson, B. Sahoo and F. Glorius, Adv. Synth. Catal., 2014, 356, 2794 CrossRef CAS; (c) X.-Z. Shu, M. Zhang, Y. He, H. Frei and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 5844 CrossRef CAS PubMed ; for selected recent examples, see: ; (d) U. A. Carrilo-Arcos and S. Porcel, Org. Biomol. Chem., 2018, 16, 1837 RSC; (e) M. Barbero and S. Dughera, Org. Biomol. Chem., 2018, 16, 295 RSC; (f) J. Xie, K. Sekine, S. Witzel, P. Krämer, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2018 DOI:10.1002/anie.201806427.
- (a) R. Cai, M. Lu, E. Y. Aguilera, Y. Xi, N. G. Akhmedov, J. L. Petersen, H. Chen and X. Shi, Angew. Chem., Int. Ed., 2015, 54, 8772 CrossRef CAS PubMed; (b) T. Cornilleau, P. Hermange and E. Fouquet, Chem. Commun., 2016, 52, 10040 RSC; (c) V. Gauchot and A.-L. Lee, Chem. Commun., 2016, 52, 10163 RSC; (d) S. Witzel, J. Xie, M. Rudolph and A. S. K. Hashmi, Adv. Synth. Catal., 2017, 359, 1522 CrossRef CAS; (e) C. Sauer, Y. Liu, A. De Nisi, S. Protti, M. Fagnoni and M. Bandini, ChemCatChem, 2017, 9, 4456 CrossRef CAS.
- For selected examples on the biaryl reductive elimination of gold complexes, see: (a) J. Vicente, M. D. Bermùdez, J. Escribano, M. P. Carrillo and P. G. Jones, J. Chem. Soc., Dalton Trans., 1990, 3083 RSC; (b) J. Vicente, M. D. Bermùdez and J. Escribano, Organometallics, 1991, 10, 3380 CrossRef CAS; (c) W. J. Wolfe, M. S. Winston and F. D. Toste, Nat. Chem., 2014, 6, 159 CrossRef PubMed; (d) K. Kang, S. Liu, T. Xu, D. Wang, X. leng, R. Bai, Y. Lan and Q. Shen, Organometallics, 2017, 36, 4727 CrossRef CAS.
- Q. Zhang, Z.-Q. Zhang, Y. Fu and H.-Z. Yu, ACS Catal., 2016, 6, 798 CrossRef CAS.
- For selected reviews, see: (a) M. C. Kozlowski, B. J. Morgan and E. C. Linton, Chem. Soc. Rev., 2009, 38, 3193 RSC; (b) P. Loxqa, E. Manourya, R. Poli, E. Deydier and A. Labande, Coord. Chem. Rev., 2016, 308, 131 CrossRef.
- For selected examples on the transmetallation of boronic acids with Au(I) species, see: (a) D. V. Partyka, M. Zeller, A. D. Hunter and T. G. Gray, Angew. Chem., Int. Ed., 2006, 45, 8188 CrossRef CAS PubMed; (b) D. V. Partyka, J. B. Updegraff III, M. Zeller, A. D. Hunter and T. G. Gray, Organometallics, 2009, 28, 1666 CrossRef CAS; (c) A. S. K. Hashmi, T. Dondeti Ramamurthi and F. Rominger, J. Organomet. Chem., 2009, 694, 592 CrossRef CAS; (d) D. V. Partyka, M. Zeller, A. D. Hunter and T. G. Gray, Inorg. Chem., 2012, 51, 8394 CrossRef CAS PubMed; (e) S. Dupuy, L. Crawford, M. Bühl, A. M. Z. Slawin and S. P. Nolan, Adv. Synth. Catal., 2012, 354, 2380 CrossRef CAS.
- For selected examples on the transmetallation of boronic acids with Au(III) species, see ref. 7d and: (a) N. P. Mankad and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 12859 CrossRef CAS PubMed; (b) M. Hofer, E. Gomez-Bengoa and C. Nevado, Organometallics, 2014, 33, 1328 CrossRef CAS.
- A. N. Cammidge and K. V. L. Crépy, Chem. Commun., 2000, 1723 RSC.
- E. Bélanger, M.-F. Pouliot and J.-F. Paquin, Org. Lett., 2009, 11, 2201 CrossRef PubMed.
- L. Wei, L. Yao, Z.-F. Wang, H. Li, H.-Y. Tao and C.-J. Wang, Adv. Synth. Catal., 2016, 358, 3748 CrossRef CAS.
- For recent examples of chiral Au(III) complexes, see: (a) J.-F. Cui, H.-M. Ko, K.-P. Shing, J.-R. Deng, N. C.-H. Lai and M.-K. Wong, Angew. Chem., Int. Ed., 2017, 56, 3074 CrossRef CAS PubMed; (b) P. T. Bohan and F. D. Toste, J. Am. Chem. Soc., 2017, 139, 11016 CrossRef CAS PubMed; (c) J. Rodriguez and D. Bourissou, Angew. Chem., Int. Ed., 2018, 57, 386 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental procedures and analysis/spectroscopic data (1H NMR, 13C NMR, 31P NMR and HPLC chromatograms). CCDC 1843202. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc07530a |
| ‡ A second minor peak was detected at 13.2 ppm by 31P NMR (<10%). The exact structure of this side-product could not be clearly determined. See the ESI† for details. |
| § Vapor diffusion method employing dichloromethane and diethyl ether as solvents. Crystallographic data for this structure have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 1843202.† |
| This journal is © The Royal Society of Chemistry 2018 |