
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegiocontrol in the cobalt-catalyzed hydrosilylation of alkynes†
Guojiao
Wu
,
Uttam
Chakraborty
and
Axel
Jacobi von Wangelin
 *
*
Department of Chemistry, University of Hamburg Martin Luther King Pl. 6, 20146 Hamburg, Germany. E-mail: axel.jacobi@uni-hamburg.de
First published on 5th October 2018
Abstract
Hydrofunctionalizations of unsaturated hydrocarbons are key strategies for the synthesis of functionalized building blocks. Here, we report highly versatile cobalt-catalyzed hydrosilylations of alkynes that operate with minute amounts of the inexpensive, bench-stable pre-catalyst Co(OAc)2·4H2O under mild conditions (0.1–1 mol%, THF, r.t., 1 h). Near-perfect regiocontrol/stereocontrol was induced by the choice of the ligand: bidentate phosphines afforded (E)-β-vinylsilanes; α-vinylsilanes formed with bipyridine ligands.
Alkenylsilanes constitute versatile building blocks in the realm of fine chemicals and materials synthesis by virtue of their dense poly-functionalization.1 The combination of a polarized alkene moiety, a Si center, and the substituents at alkene and silicon offer ample opportunities for post-synthesis manipulations. Hydro-silylations of alkynes enable a most straightforward and atom-economic synthesis of alkenylsilanes in the presence of the noble metal catalysts Ru, Rh, and Pt.2–4 Only few protocols rely on the use of inexpensive and environmentally benign 3d base metal catalysts.5 Despite the recent progress in the field, the precise control of regioselectivity and stereoselectivity remains a challenge of utmost importance. Cobalt catalysts were demonstrated to exhibit especially high activity and tolerance of functional groups in hydrosilylations of alkenes.6 Much less attention has been directed towards cobalt-catalyzed hydrosilylations of alkynes which often require high catalyst loadings, complex ligands, and harsh conditions or showed poor regio/stereocontrol or a limited substrate scope with regard to alkynes and silanes (Scheme 1, top).7–12 Very recently, Ge et al. reported Co-catalyzed hydrosilylations to give (Z)-vinylsilanes in the presence of pyridine-2,6-diimines.13 We believed that a most user-friendly protocol would combine the following criteria: (i) high catalytic activity of a commercial catalyst system under very mild conditions; (ii) control of regioselectivity and stereoselectivity by the choice of the ligand, and (iii) a wide substrate scope involving terminal and internal alkynes and trihydrosilanes. Documented herein are the benefits of a versatile regiodivergent and stereoselective hydrosilylation of alkynes in the presence of only 0.1–1 mol% Co(OAc)2·4H2O and commercial phosphine or bipyridine ligands (Scheme 1, bottom).
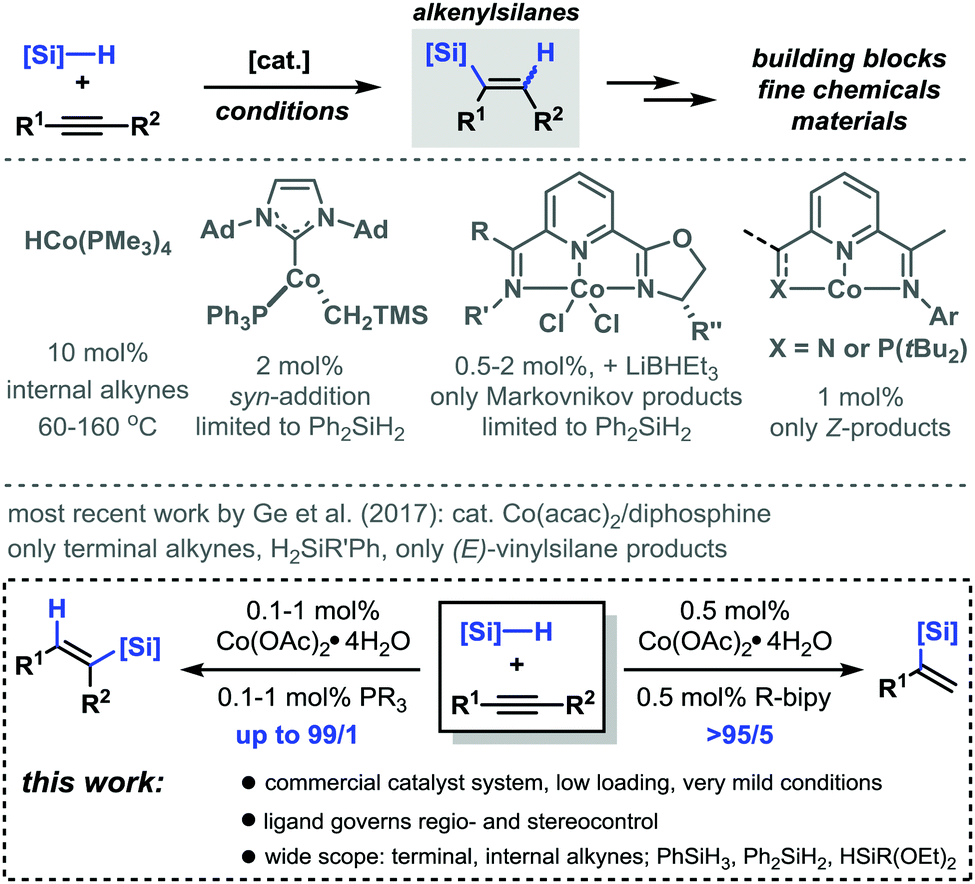 | ||
| Scheme 1 Cobalt-catalyzed hydrosilylations of alkynes. | ||
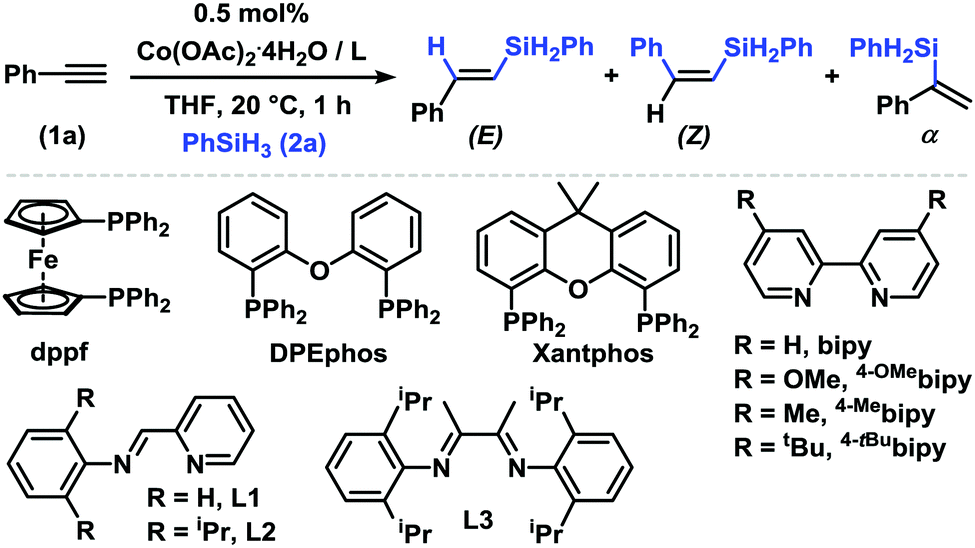
An initial evaluation of parameters in the model reaction between phenylacetylene (1a) and phenylsilane (2a) in the presence of the bench-stable and inexpensive Co(OAc)2·4H2O revealed very good regioselectivity and stereoselectivity toward (E)-styrylsilane with various commercial phosphine ligands (Table 1, entries 1–7). With only 0.1 mol% Co(OAc)2·4H2O/dppb, an isolated yield of 86% was obtained with very high stereoselectivity (>50/1 E/Z) and regiocontrol (1/49 α/β). A complete reversal of regioselectivity was observed upon employment of bipyridine ligands (up to 25/1 α/β, entries 8–12). These results are a significant extension of previous reports with N,N,N-ligands that resulted in poor regio-selectivity with PhSiH3.12 The strict ligand control of this protocol is further documented by the lack of catalytic activity in the presence of other N,N-ligands such as (pyridin-2-yl)methanimine, butane-2,3-diimine, and terpyridine (entries 13 and 14).
|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Yieldb [%] | E/Z/αb | |
| a Conditions: 1a (0.40 mmol), 2a (0.48 mmol), Co(OAc)2·4H2O (0.5 mol%), ligand (0.5 mol%), 0.5 mL THF, 20 °C, under N2 (1 h w/PR3, 3 h with N,N-ligands). b Yield and product ratios from quantitative GC-FID vs. internal n-dodecane. c 0.1 mol% Co(OAc)2·4H2O (in 10 μL methanol), 0.1 mol% dppb, 1 h. d Isolated yields in parentheses. | ||||
| 1 |

|
n = 1 | 72 | 87/0/13 |
| 2 | n = 2 | 81 | 97/0/3 | |
| 3 | n = 3 | 95 | 98/0/2 | |
| 4 | Dppf | 92 | 97/0/3 | |
| 5 | DPEphos | 90 | 98/0/2 | |
| 6 | Xantphos | 89 | 87/2/11 | |
| 7 | Dppb | 94 (86) | 98/0/2 | |
| 8 | 2,2′-Bipyridine (bipy) | 75 | 10/0/90 | |
| 9 | 4-OMebipy | 66 | 4/0/96 | |
| 10 | 4-Me bipy | 70 (60) | 5/0/95 | |
| 11 | 4-tBubipy | 64 | 5/0/95 | |
| 12 | 1,10-Phenanthroline | 61 | 10/0/90 | |
| 13 | L1 or L2 or L3 | 0 | — | |
| 14 | 2,2′;6′,2′′-Terpyridine | 0 | — | |
Various arylacetylenes underwent clean formation of (E)-alkenyl-silanes in good yields, very high stereoselectivities (>50/1 E/Z) and regioselectivities (>18/1 β/α) with only 0.1 mol% Co(OAc)2·4H2O and dppb at r.t. (Scheme 2). Diverse substitution patterns (incl. ortho-substituents) and functional groups (OH, NH2, nitrile, ester, aldehyde, thiophene, and pyridine) were tolerated. No dehalogenation was observed with halides (Br, Cl, and F). The same conditions were successfully applied to hydrosilylations with monohydrosilanes and dihydrosilanes (i.e. (EtO)3SiH and Ph2SiH2). An extension of the methodology to terminal and internal alkyl alkynes was realized with cobalt/diphosphine catalyst systems (entries 5 and 6 in Table 1 and ESI†). Terminal alkynes exhibited the highest reactivities, very high stereoselectivities (>50/1 E/Z), and very high regioselectivities (up to 99/1 β/α) toward (E)-alkenyl-silanes in the presence of 1 mol% Co(OAc)2·4H2O and DPEphos (Scheme 3, top). Silylethers, halides, nitriles, and ester moieties were tolerated. Free OH groups inhibited the conversion. The protocol was also applied to hydrosilylation with diphenylsilane (5m).
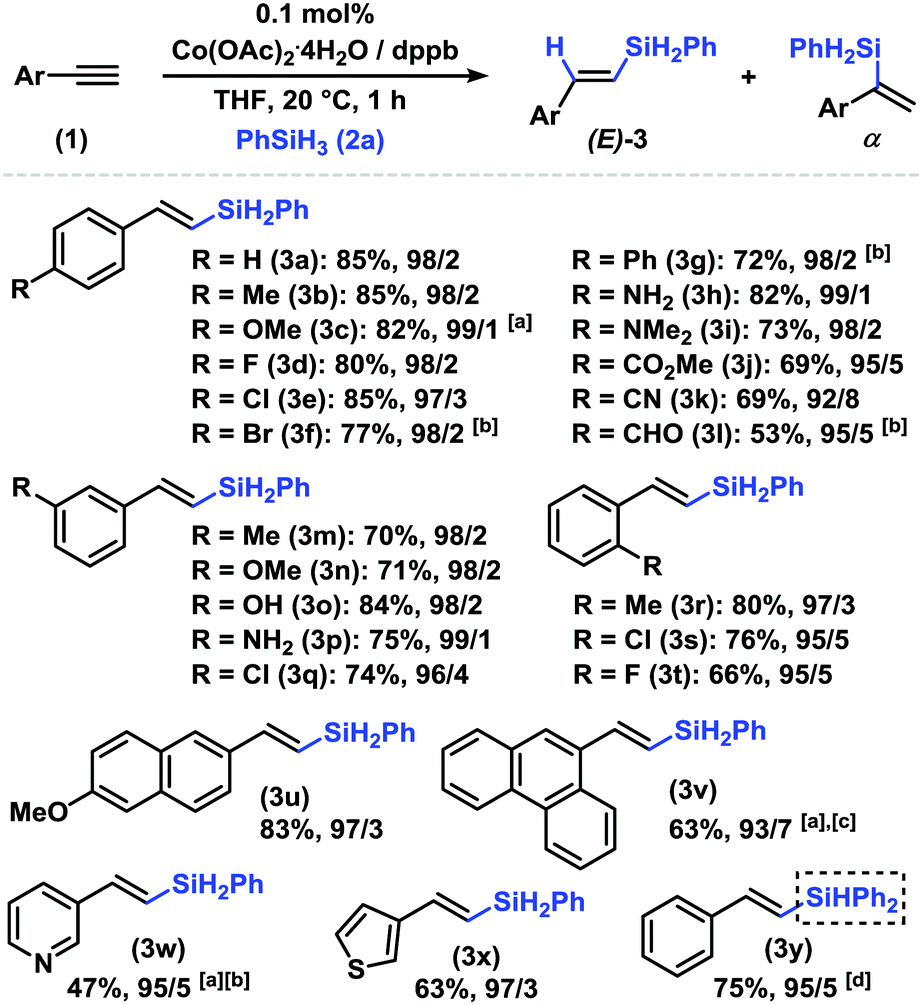 | ||
Scheme 2 Hydrosilylation of terminal aryl alkynes. Conditions: 1 (0.4 mmol), 2a (0.48 mmol), Co(OAc)2·4H2O (0.1 mol%), dppb (0.1 mol%), 0.5 mL THF, 20 °C, under N2, 1 h. Isolated yields are given. E/α ratios were determined by quantitative GC-FID vs. internal n-dodecane. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3 h. bCo(OAc)2·4H2O (0.5 mol%), dppb (0.5 mol%). cCo(OAc)2·4H2O (1 mol%), dppb (1 mol%). dXantphos. 3 h. bCo(OAc)2·4H2O (0.5 mol%), dppb (0.5 mol%). cCo(OAc)2·4H2O (1 mol%), dppb (1 mol%). dXantphos. | ||
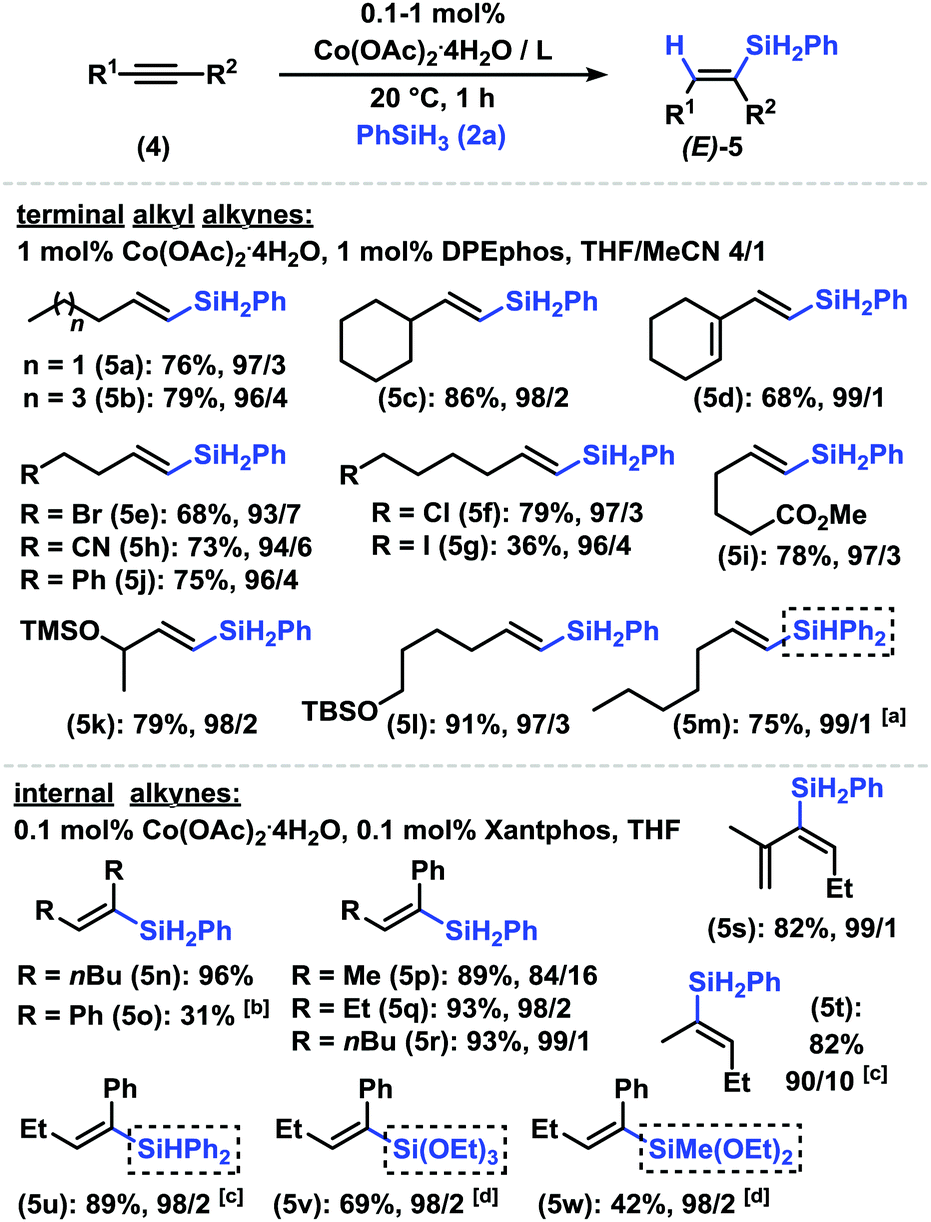 | ||
| Scheme 3 Hydrosilylation of alkynes. Conditions: 4 (0.4 mmol), 2a (0.48 mmol), Co(OAc)2·4H2O (0.1 or 1 mol%), ligand (0.1 or 1 mol%), 0.5 mL solvent, 1 h, under N2. Isolated yields are given. Product ratios E/α were determined by quantitative GC-FID vs. internal n-dodecane. aXantphos. bCo(OAc)2·4H2O (0.5 mol%). cCo(OAc)2·4H2O (1 mol%). dCo(OAc)2·4H2O (0.5 mol%), 0.5 mL MeCN, 60 °C, 2 h. | ||
Internal alkynes successfully reacted under slightly modified conditions with Xantphos as a ligand (Scheme 3, bottom). Highly selective syn-hydrosilylation was operative with all substrates. Unsymmetrical alkynes engaged in the regioselective addition of the silyl moiety to the less bulky C atom (Ph, i-Pr vs. alkyl; alkyl vs. Me). This is also manifested in the series of 2-alkyl phenylacetylenes with increasing regioselectivities in the order Me < Et < nBu (5p, 5q, and 5r). The conjugated enyne 2-methylhex-1-ene-3-yne cleanly afforded the desired 3-silyl product 5s. The hydrosilylation of the sterically rather unbiased 2-pentyne gave impressive regio-selectivity (9/1) and stereoselectivity. Ph2SiH2, HSi(OEt)3, and HSiMe(OEt)2 fared equally well. Steric silanes (HSiEt3 and HSi(OiPr)Me2) gave complex product mixtures, possibly from rapid alkyne (cyclo)oligomerizations. We further explored the regioselective α-silylation of terminal alkynes (entries 8–12, Table 1). The Co/2,2′-bipyridine catalysts enabled a reversal of regio-selectivity to cleanly afford 1-phenylvinyl silanes which constitute important synthetic building blocks (Scheme 4).1 The reaction displayed compatibility with Br, NH2, ester, nitrile, and free OH functional groups. 2-Ethynyl-6-methoxynaphthalene, 3-ethynylthiophene, and 3-ethynylpyridine gave slightly lower conversions (6n, 6o, and 6p). Internal alkynes reacted poorly (∼10% yield, low regiocontrol).
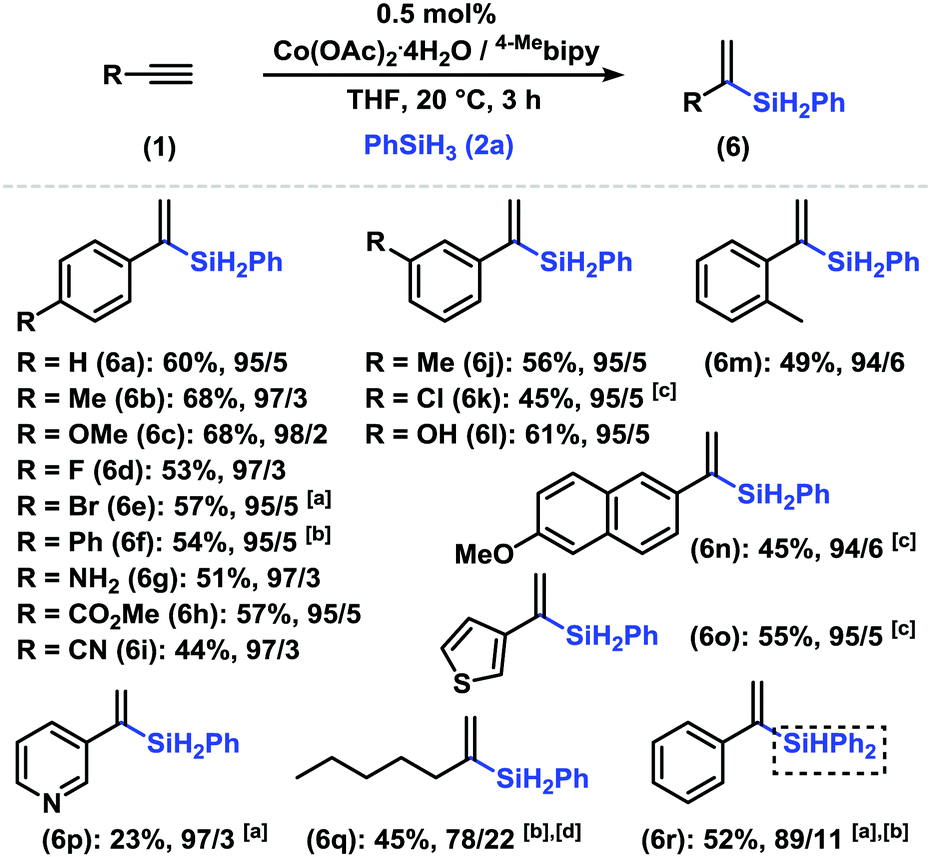 | ||
| Scheme 4 Markovnikov hydrosilylation of arylacetylenes. Conditions: 1 (0.4 mmol), 2a (0.48 mmol), Co(OAc)2·4H2O (0.1 or 1 mol%), ligand (0.1 or 1 mol%), 0.5 mL solvent for 1 h under N2. Isolated yields are given. The product ratios [α/E] were determined by GC analysis. a50 °C. b6 h. c2a (1.5 equiv.). d4-OMebipy as a ligand (0.5 mol%). | ||
The versatility of the derived alkenylsilanes for further manipulations is exemplified in Scheme 5. Sequential hydro-silylations afforded a divinylsilane (87/13 β/α) via the alkenyl-silane 3a. Substitution of the hydride at Si with Grignard reagents is a robust method of silane functionalization. With PhMgBr, tertiary silane 3a was obtained in 74% yield. Tamao oxidation of vinylsilane 6a gave the corresponding phenone in 81% yield. Further alkenylsilane reactions of high utility include electrophilic and nucleophilic olefin additions, silyl substitutions, oxidations, cross-couplings, hydrofunctionalizations and polymerizations.1
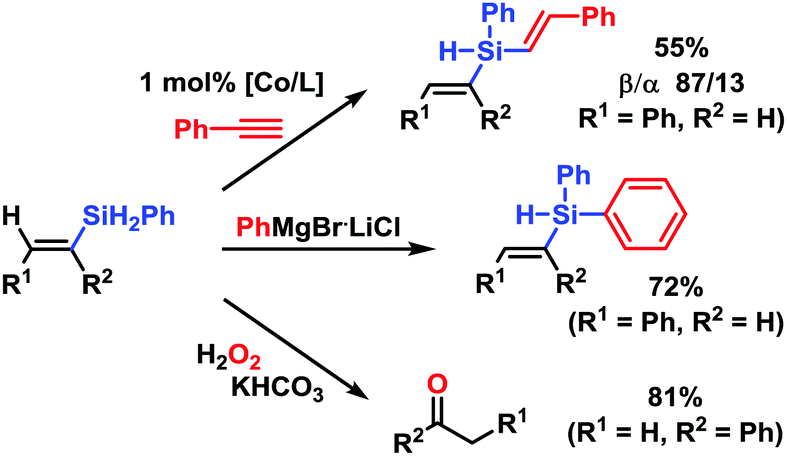 | ||
| Scheme 5 Post-synthesis manipulations of (E)-alkenyl silanes. | ||
In extension of literature precedents and our own preliminary mechanistic data derived from the optimization experiments, substrate scope, and regiochemical and stereochemical course of the cobalt-catalyzed hydrosilylation reactions, we performed key mechanistic studies on the nature of catalytic intermediates (Scheme 6).6,10–15 In full accord with the literature,6g the reduction of Co(OAc)2 by the silane in the presence of diphosphine ligands (L = P2) was observed by MS and NMR. We postulate the formation of the monohydrido species LnCoH (A-I). Indeed, the penta-coordinate complex (dppb)2CoH was observed in LIFDI-MS spectra (m/z 912.27) and showed a characteristic 1H NMR resonance at −14.5 ppm (see the ESI† for details). Coordination of the alkyne (A-II) and migratory insertion into the Co–H bond constitute the elemental steps that govern the regioselectivity and stereoselectivity of the reaction. The preferential addition of the cobalt complex to the less hindered side of the alkyne bearing the smaller substituent Rs affords the more stable alkenylcobalt species A-III. Formal trans-metalation to Si results in the formation of the (E)-alkenylsilane and regeneration of the active species A-I. The reaction is first order in the cobalt catalyst and zero order in phenylacetylene and silane (see the ESI†), which suggests that the alkyne insertion into Co–H is rate-determining. A different mechanistic scenario appears to be operative with bipy ligands (L = N2, Scheme 6, bottom). The reductive formation of a silylcobalt complex LCo-Si (B-I) is in full agreement with the literature.6c,6f,111H NMR spectra of the reaction of Co(OAc)2·4H2O/4Mebipy with PhSiH3 (1:1:10) documented the anticipated formation of a paramagnetic species. The presence of silylcobalt complexes was suggested by LIFDI-MS measurements of the catalyst mixture which exhibited the trisilyl complex (4Mebipy)2Co(SiHPhSiHPhSiH2Ph) (m/z 746.00). Such oligosilane complexes constitute key intermediates in silane dehydro-coupling and oligomerization reactions and were also observed with other metals.15 The same paramagnetic oligosilyl complex was independently formed by the reaction of equimolar Co(OAc)2·4H2O and 4-Mebipy with 5 equiv. of PhSiH3. The dehydrocoupling could be reversed by the addition of LiAlH4 (2.5 equiv. per [Co]) which resulted in the generation of PhSiH3 (see the ESI† for details). The silylcobalt complex B-I is postulated to engage in alkyne coordination followed by regioselective and stereo-selective 1,2-syn-insertion. The resultant syn-alkenylcobalt complex B-III releases α-alkenylsilane upon reaction with PhSiH3. This hydrosilylation reaction is first order in [Co] and silane and zero order in phenylacetylene (see the ESI†). This indicates a rate limitation by the product release step.
 | ||
| Scheme 6 Proposed reaction mechanisms of hydrosilylation catalysed by (diphosphine)cobalt complexes (top) and (bipy)cobalt complexes (bottom). | ||
In conclusion, a highly versatile cobalt-catalyzed hydrosilylation has been developed that enables precise regiocontrol by the choice of the ligand. The catalysts exhibit superior activity over the current state-of-the-art, operating under very mild conditions (20 °C, 1 h) with only 0.1–1 mol% catalyst loading. The catalysts are based on commercial and inexpensive components: the bench-stable Co(OAc)2·4H2O and the ligand dppb or 4-Mebipy. The mild conditions allow a wide substrate scope (terminal and internal alkynes, various silanes) and the tolerance of sensitive functional groups (halides, aldehydes, esters, nitriles, NH2, and OH). Key mechanistic studies support the notion of a mechanistic dichotomy: the ligand dppb enables highly selective formation of (E)-alkenyl-silanes via anti-Markovnikov hydrosilylation. A full regiochemical switch is effected by the ligand 4-Mebipy which selectively delivers Markovnikov products. The former pathway involves the formation of hydridocobalt catalyst species, while the latter mode of reactivity is most likely based on silylcobalt species. The high functional group tolerance and mild reaction conditions make these protocol highly attractive for complex molecule synthesis with great utility for medicinal and materials chemistry endeavours.
This work was supported by the Deutsche Forschungsgemeinschaft (JA 1107/6-1) and the European Research Council (CoG 683150).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) E. Langkopf and D. Schinzer, Chem. Rev., 1995, 95, 1375 CrossRef CAS; (b) I. Fleming, A. Barbero and D. Walter, Chem. Rev., 1997, 97, 2063 CrossRef CAS PubMed; (c) I. Ojima, Z. Li and J. Zhu, The Chemistry of Organic Silicon Compounds, Wiley, Hoboken, 2003, pp. 1687 Search PubMed; (d) S. E. Denmark and R. F. Sweis, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, Weinheim, 2nd edn, 2008, pp. 163 Search PubMed.
- (a) C. S. Arico and L. R. Cox, Org. Biomol. Chem., 2004, 2, 2558 RSC; (b) S. V. Maifeld, M. N. Tran and D. Lee, Tetrahedron Lett., 2005, 46, 105 CrossRef CAS; (c) M. Nagao, K. Asano, K. Umeda, H. Katayama and F. Ozawa, J. Org. Chem., 2005, 70, 10511 CrossRef CAS PubMed; (d) S. Ding, L. Song, L. W. Chung, X. Zhang, J. Sun and Y. D. Wu, J. Am. Chem. Soc., 2013, 135, 13835 CrossRef CAS PubMed; (e) R. Gao, D. R. Pahls, T. R. Cundari and C. S. Yi, Organometallics, 2014, 33, 6937 CrossRef CAS; (f) S. Ding, L. J. Song, Y. Wang, X. Zhang, L. W. Chung, Y. D. Wu and J. Sun, Angew. Chem., Int. Ed., 2015, 54, 5632 CrossRef CAS PubMed; (g) Y. Mutoh, Y. Mohara and S. Saito, Org. Lett., 2017, 19, 5204 CrossRef CAS PubMed.
- (a) A. Mori, E. Takahisa, Y. Yamamura, T. Kato, A. P. Mudalige, H. Kajiro, K. Hirabayashi, Y. Nishihara and T. Hiyama, Organometallics, 2004, 23, 1755 CrossRef CAS; (b) G. T. S. Andavan, E. B. Bauer, C. S. Letko, T. K. Hollis and F. S. Tham, J. Organomet. Chem., 2005, 690, 5938 CrossRef CAS; (c) T. Sanada, T. Kato, M. Mitani and A. Mori, Adv. Synth. Catal., 2006, 348, 51 CrossRef CAS; (d) J. P. Morales-Cerón, P. Lara, J. López-Serrano, L. L. Santos, V. Salazar, E. Álvarez and A. Suárez, Organometallics, 2017, 36, 2460 CrossRef.
- (a) M. Chauhan, B. J. Hauck, L. P. Keller and P. Boudjouk, J. Organomet. Chem., 2002, 645, 1 CrossRef CAS; (b) W. Wu and C.-J. Li, Chem. Commun., 2003, 1668 RSC; (c) H. Aneetha, W. Wu and J. G. Verkade, Organometallics, 2005, 24, 2590 CrossRef CAS; (d) A. Hamze, O. Provot, J.-D. Brion and M. Alami, Tetrahedron Lett., 2008, 49, 2429 CrossRef CAS; (e) A. Hamze, O. Provot, J.-D. Brion and M. Alami, J. Organomet. Chem., 2008, 693, 2789 CrossRef CAS; (f) G. Berthon-Gelloz, J.-M. Schumers, G. D. Bo and I. E. Markó, J. Org. Chem., 2008, 73, 4190 CrossRef CAS PubMed; (g) J. Hu, F. Li and T. S. A. Hor, Organometallics, 2009, 28, 1212 CrossRef CAS; (h) Y. Kawasaki, Y. Ishikawa, K. Igawa and K. Tomooka, J. Am. Chem. Soc., 2011, 133, 20712 CrossRef CAS PubMed; (i) R. Cano, M. Yus and D. J. Ramón, ACS Catal., 2012, 2, 1070 CrossRef CAS.
- For Fe, see: (a) C. Belger and B. Plietker, Chem. Commun., 2012, 48, 5419 RSC; (b) M. D. Greenhalgh, D. J. Frank and S. P. Thomas, Adv. Synth. Catal., 2014, 356, 584 CrossRef CAS; (c) J. H. Docherty, J. Peng, A. P. Dominey and S. P. Thomas, Nat. Chem., 2017, 9, 595 CrossRef CAS PubMed . For Ni catalysts, see: ; (d) K. Tamao, M. Asahara and A. Kawachi, J. Organomet. Chem., 1996, 521, 325 CrossRef CAS; (e) A. Tillack, S. Pulst, W. Baumann, H. Baudisch, K. Kortus and U. Rosenthal, J. Organomet. Chem., 1997, 532, 117 CrossRef CAS; (f) M. J. Chaulagain, G. M. Mahandr and J. Montgomery, Tetrahedron Lett., 2006, 62, 7560 CrossRef CAS; (g) J. Berding, J. A. Van Paridon, V. H. S. Van Rixel and E. Bouwman, Eur. J. Inorg. Chem., 2011, 2450 CrossRef CAS . For Co catalysts, see ref. 8–14.
- For selected examples: (a) Z. Mo, Y. Liu and L. Deng, Angew. Chem., Int. Ed., 2013, 52, 10845 CrossRef CAS PubMed; (b) C. Chen, M. B. Hecht, A. Kavara, W. W. Brennessel, B. Q. Mercado, D. J. Weix and P. L. Holland, J. Am. Chem. Soc., 2015, 137, 13244 CrossRef CAS PubMed; (c) X. Du, Y. Zhang, D. Peng and Z. Huang, Angew. Chem., Int. Ed., 2016, 55, 6671 CrossRef CAS PubMed; (d) D. Noda, A. Tahara, Y. Sunada and H. Nagashima, J. Am. Chem. Soc., 2016, 138, 2480 CrossRef CAS PubMed; (e) C. H. Schuster, T. Diao, I. Pappas and P. J. Chirik, ACS Catal., 2016, 6, 2632 CrossRef CAS; (f) A. D. Ibrahim, S. W. Entsminger, L. Zhu and A. R. Fout, ACS Catal., 2016, 6, 3589 CrossRef CAS; (g) C. Wang, W. J. Teo and S. Ge, ACS Catal., 2017, 7, 855 CrossRef CAS; (h) Y. Liu and L. Deng, J. Am. Chem. Soc., 2017, 139, 1798 CrossRef CAS PubMed; (i) K. L. Lee, Angew. Chem., Int. Ed., 2017, 56, 3665 CrossRef CAS PubMed; (j) B. Raya, S. Jing, V. Balasanthiran and T. V. RajanBabu, ACS Catal., 2017, 7, 2275 CrossRef CAS PubMed ; for review, see ; (k) J. Su and L. Deng, ACS Catal., 2016, 6, 290–300 CrossRef.
- T. Konno, K. Taku, S. Tamada, K. Moriyasu and T. Ishihara, Org. Biomol. Chem., 2009, 7, 1167 RSC.
- (a) M. Isobe, R. Nishizawa, T. Nishikawa and K. Yoza, Tetrahedron Lett., 1999, 40, 6927 CrossRef CAS; (b) S. Tojo and M. Isobe, Tetrahedron Lett., 2005, 46, 381 CrossRef CAS; (c) K.-H. Huang and M. Isobe, Eur. J. Org. Chem., 2014, 4733 CrossRef CAS.
- L. Yong, K. Kirleis and H. Butenschön, Adv. Synth. Catal., 2006, 348, 833 CrossRef CAS.
- Z. Mo, J. Xiao, Y. Gao and L. Deng, J. Am. Chem. Soc., 2014, 136, 17414 CrossRef CAS PubMed.
- (a) J. Guo and Z. Lu, Angew. Chem., Int. Ed., 2016, 55, 10835 CrossRef CAS PubMed; (b) Z. Zuo, J. Yang and Z. Huang, Angew. Chem., Int. Ed., 2016, 55, 10839 CrossRef CAS PubMed; (c) J. Guo and Z. Lu, Angew. Chem., Int. Ed., 2017, 56, 615 CrossRef CAS PubMed.
- A. Rivera-Hernández, B. J. Fallon, S. Ventre, C. Simon, M.-H. Tremblay, G. Gontard, E. Derat, M. Amatore, C. Aubert and M. Petit, Org. Lett., 2016, 18, 4242 CrossRef PubMed.
- (a) W. J. Teo, C. Wang, Y. W. Tan and S. Ge, Angew. Chem., Int. Ed., 2017, 56, 4328 CrossRef CAS PubMed; (b) X. Du, W. Hou, Y. Zhang and Z. Huang, Org. Chem. Front., 2017, 4, 1517 RSC; (c) C. Wu, W. J. Teo and S. Ge, ACS Catal., 2018, 8, 5896 CrossRef CAS.
- C. C. H. Atienza, T. Diao, K. J. Weller, S. A. Nye, K. M. Lewis, J. G. P. Delis, J. L. Boyer, A. K. Roy and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 12108 CrossRef CAS PubMed.
- (a) M. D. Spencer, Q. D. Shelby and G. S. Girolami, J. Am. Chem. Soc., 2007, 129, 1860 CrossRef CAS PubMed; (b) E. E. Smith, G. Du, P. E. Fanwick and M. M. Abu-Omar, Organometallics, 2010, 29, 6527 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc07267a |
| This journal is © The Royal Society of Chemistry 2018 |