
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA supramolecular porous material comprising Fe(II) mesocates†
Benjamin H.
Wilson
a,
Hayley S.
Scott
a,
Omid T.
Qazvini
b,
Shane G.
Telfer
 b,
Corine
Mathonière
cd,
Rodolphe
Clérac
ef and
Paul E.
Kruger
*a
b,
Corine
Mathonière
cd,
Rodolphe
Clérac
ef and
Paul E.
Kruger
*a
aMacDiarmid Institute for Advanced Materials and Nanotechnology, School of Physical and Chemical Sciences, University of Canterbury, Private Bag 4800, Christchurch 8041, New Zealand. E-mail: paul.kruger@canterbury.ac.nz
bMacDiarmid Institute for Advanced Materials and Nanotechnology, Institute of Fundamental Sciences, Massey University, Palmerston North 4442, New Zealand
cCNRS, ICMCB, UMR 5026, F-33600 Pessac, France
dUniv. Bordeaux, ICMCB, UMR 5026, F-33600 Pessac, France
eCNRS, CRPP, UMR 5031, F-33600 Pessac, France
fUniv. Bordeaux, CRPP, UMR 5031, F-33600 Pessac, France
First published on 7th November 2018
Abstract
The dinuclear mesocate [Fe2L3](BF4)4, 1, is a supramolecular building block for a microporous material. Structural analysis reveals that extensive noncovalent interactions in the solid state generate a 3D framework with microporous channels. These channels are permanently accessible to incoming guest molecules and adsorption isotherms demonstrate that the material has a high selectivity for CO2 over N2.
Over the past two decades, our understanding of microporous materials has progressed, steadily driven by their relevance to diverse applications such as gas separation and storage, catalysis, small-molecule separation and chemical sensing.1 3D polymeric architectures, sustained by covalent and metal–ligand interactions, comprise the majority of microporous materials that have uniform and periodic structures. Notably, these include naturally occurring zeolites,2 metal–organic-frameworks (MOFs)3 and covalent-organic-frameworks (COFs).4 More recently, however, an increasing number of studies have been devoted to porous materials comprised of discrete molecular entities.5 This class of compound, called molecular porous materials (MPMs),6 offer synthetic advantages over 3D polymeric materials owing to their enhanced solubility, which may aid their processability and incorporation into functional materials (e.g. attachment to surfaces, etc.). Two broad classes, depending on their structural conformation, describe the porosity within MPMs. Intrinsic porosity refers to compounds in which accessible void space is found inside a discrete entity e.g. organic cages,7 metal–organic-squares (MOSs)8 and polyhedra (MOPs)9etc. Extrinsic porosity refers to structures in which void space exists between molecular units. Because the packing density in conventional molecule-based solids is high, the formation of MPMs possessing extrinsic porosity is rare. However, a reduction of the packing density via constructive and supportive supramolecular interactions between molecules can realise porous architectures. These supramolecular interactions typically involve hydrogen bonding, as in hydrogen-bonded organic frameworks (HOFs),10 supramolecular metal–organic frameworks (SMOFs),11 and supramolecular organic frameworks (SOFs),12 or noncovalent interactions involving strong dipoles.5a,12 There is much to learn from new examples of extrinsically porous materials, in particular the relationship between structural features and their properties, including stability, permanent porosity and solubility.
As part of our programme investigating Fe(II) dinuclear complexes13 (Fig. 1), we report here a new and rare example of an extrinsically porous material comprising a supramolecular building block. The compound, [Fe2L3](BF4)4, 1, where L = N,N-3-bis[1-(2-pyrazinyl)ethylidene]isophthalo-hydrazide, was characterised using single crystal X-ray diffraction, and its permanent porosity was evaluated by gas adsorption isotherms using a range of adsorbates.
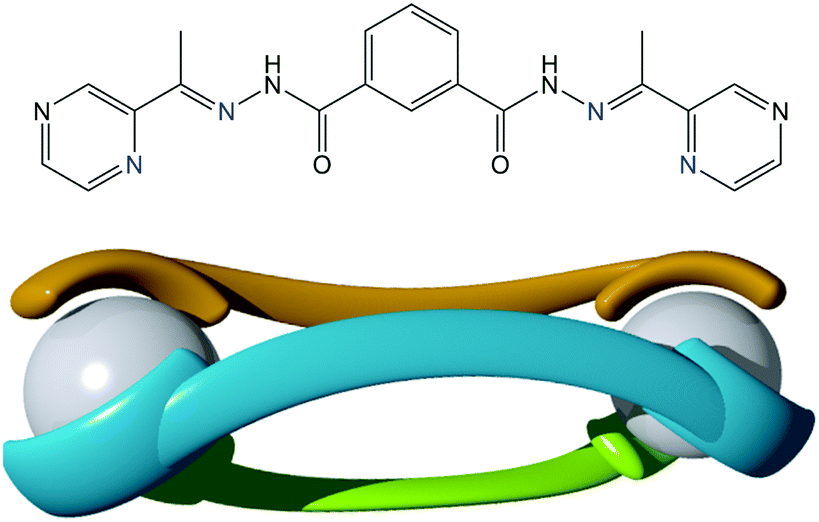 | ||
| Fig. 1 (top) Structure of the ligand, L, used in the current study with coordinating N-atoms coloured blue. (bottom) Schematic of a dinuclear triply stranded mesocate of composition [M2L3]n+. | ||
A red coloured solution resulted when a suspension of L in nitromethane was stirred with Fe(BF4)2 (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) at room temperature overnight. Single crystals of 1 were obtained by the diffusion of toluene anti-solvent into the nitromethane solution (see ESI†). The structure of 1 was determined from X-ray diffraction data collected at 120 K. It crystallises in the hexagonal space group P63/m. The asymmetric unit contains one third of an Fe(II) atom coordinated to half of a ligand fragment featuring bidentate coordination via pyrazine (N2) and hydrazide (N3) nitrogen donors (Fig. 2 and Fig. S2, ESI†). The average Fe–N bond length of 1.959(4) Å reveals the Fe(II) centre exists in the low spin state at 120 K.14 The action of a 3-fold rotoinversion congruent with the crystallographic c-axis and a reflection about a mirror plane perpendicular to the crystallographic c-axis yields the dinuclear complex, [Fe2L3]4+ (Fig. 2). The three bis-bidentate ligand strands bridge Fe(II) centres to form a compact mesocate replete with attractive noncovalent inter-strand interactions. The hydrazide oxygen atoms (O13) are twisted with respect to the plane of the central phenyl rings to which they are attached and are in close contact with an adjacent hydrazide group from a neighbouring (inter-strand) ligand, O13⋯H11(N11), 2.957(3) Å, 125.8°. Likewise, H15 from the central phenyl rings participate in an edge-to-face C–H⋯π interaction with an adjacent ligand strand, C15–H15⋯π(centre-of-ring) 2.903 Å, 149.6° (Fig. 2, Fig. S3; Tables S2, S3, ESI†). These interactions likely support the mesocate conformation.
:3) at room temperature overnight. Single crystals of 1 were obtained by the diffusion of toluene anti-solvent into the nitromethane solution (see ESI†). The structure of 1 was determined from X-ray diffraction data collected at 120 K. It crystallises in the hexagonal space group P63/m. The asymmetric unit contains one third of an Fe(II) atom coordinated to half of a ligand fragment featuring bidentate coordination via pyrazine (N2) and hydrazide (N3) nitrogen donors (Fig. 2 and Fig. S2, ESI†). The average Fe–N bond length of 1.959(4) Å reveals the Fe(II) centre exists in the low spin state at 120 K.14 The action of a 3-fold rotoinversion congruent with the crystallographic c-axis and a reflection about a mirror plane perpendicular to the crystallographic c-axis yields the dinuclear complex, [Fe2L3]4+ (Fig. 2). The three bis-bidentate ligand strands bridge Fe(II) centres to form a compact mesocate replete with attractive noncovalent inter-strand interactions. The hydrazide oxygen atoms (O13) are twisted with respect to the plane of the central phenyl rings to which they are attached and are in close contact with an adjacent hydrazide group from a neighbouring (inter-strand) ligand, O13⋯H11(N11), 2.957(3) Å, 125.8°. Likewise, H15 from the central phenyl rings participate in an edge-to-face C–H⋯π interaction with an adjacent ligand strand, C15–H15⋯π(centre-of-ring) 2.903 Å, 149.6° (Fig. 2, Fig. S3; Tables S2, S3, ESI†). These interactions likely support the mesocate conformation.
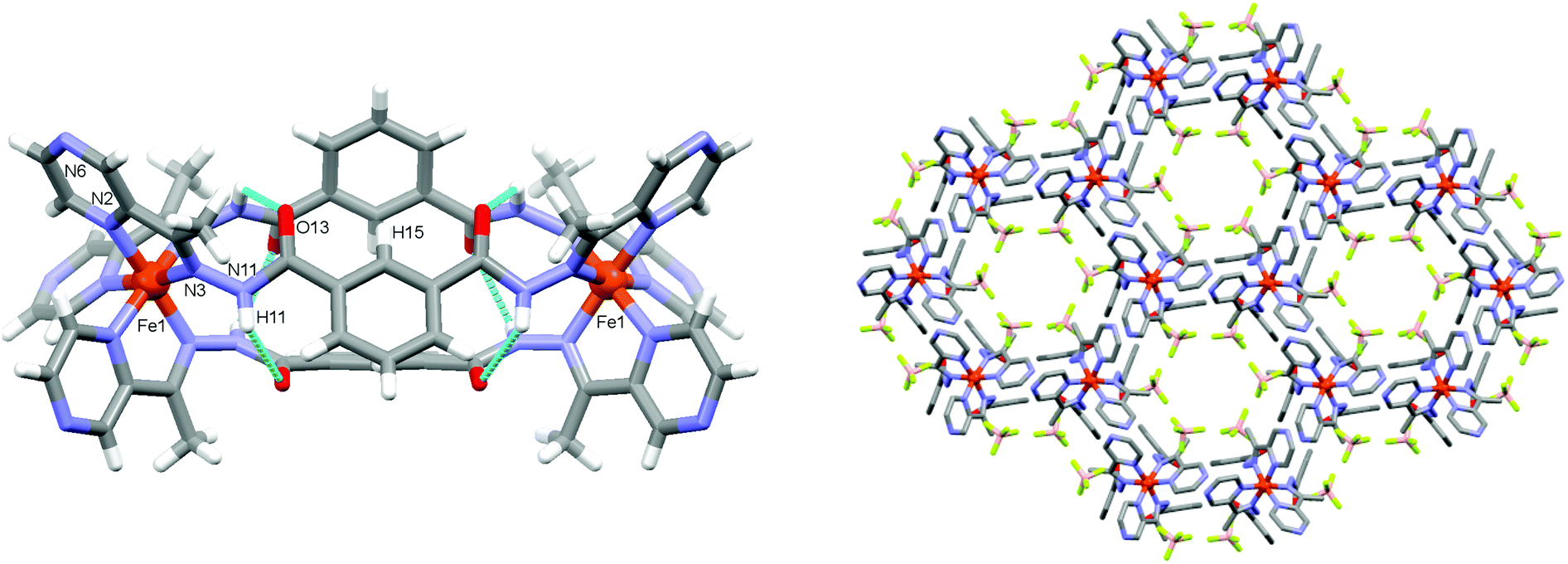 | ||
| Fig. 2 (left) Molecular and crystal structure of the dinuclear mesocate complex, 1. Anions and disordered solvate molecules removed for clarity. (right) Packing diagram of 1 view down the crystallographic c-axis. H-atoms and disordered solvate molecules omitted for clarity. | ||
The mesocates pack in the crystal lattice via extensive supramolecular interactions, the most salient of which is the hydrogen-bonding between pyrazine and hydrazide groups of adjacent mesocates e.g. N11–H11⋯N6, 3.009(8) Å, (Fig. S4, ESI†). There are six (reciprocal) hydrogen bonds between adjacent pyrazine and hydrazide moieties of neighbouring supra-molecules, along with offset face-to-face π–π interactions involving each pyrazine ring (centroid⋯centroid, 3.687(3) Å). These interactions propagate down the crystallographic c-axis to generate a network containing 1D hexagonal channels parallel with the c-axis (Fig. 2, Fig. S4–S7; Tables S2, S3, ESI†). The channel diameter at closest contact between van der Waals surfaces is ca. 5.8 Å. The tetrafluoroborate anions (which are disordered over two positions) line these 1D channels. Because of the high symmetry of the space group and disorder within the solvent molecules that reside within the channels, their identities could not be accurately determined and they were therefore treated by using the SQUEEZE programme embedded within PLATON.15 The solvent accessible void was calculated to be 1164 Å3 and accounts for 26% of the crystal volume.
Intrigued by the presence and size of the 1D channels within this material, and the structural resilience of the crystals when removed from their mother liquor, we set about to appraise its permanent porosity. Powder X-ray diffraction (PXRD) verified that activation of a batch of crystals via solvent exchange and drying in vacuo successfully retained the lattice structure (Fig. S8, ESI†). The accessibility of the activated material to incoming guests molecules was confirmed by a H2 adsorption isotherm at 77 K, which showed an uptake in excess of 60 cm3 g−1 (Fig. S9, ESI†). Hysteresis is also evident in this isotherm and is most probably a consequence of the small size of the pores and their one dimensional nature, which leads to slow diffusion of H2 at 77 K. Consistent with the small size of the 1D channels, the uptake of N2 at 77 K was hampered by slow diffusion kinetics, which precluded the measurement of a conventional BET surface area. Gas uptake around room temperature was unhindered, however, and adsorption isotherms at 273 and 295 K were measured for the adsorbates CO2, O2, N2, CH4, C2H4 and C2H6 (Fig. 3, Table 1 and Fig. S14, ESI†). The small hysteresis in the 295 K isotherms for ethane possibly arises as a consequence of its larger size and indicates that we are approaching the size limit of the guests that can be adsorbed into the small 1D channels in 1.
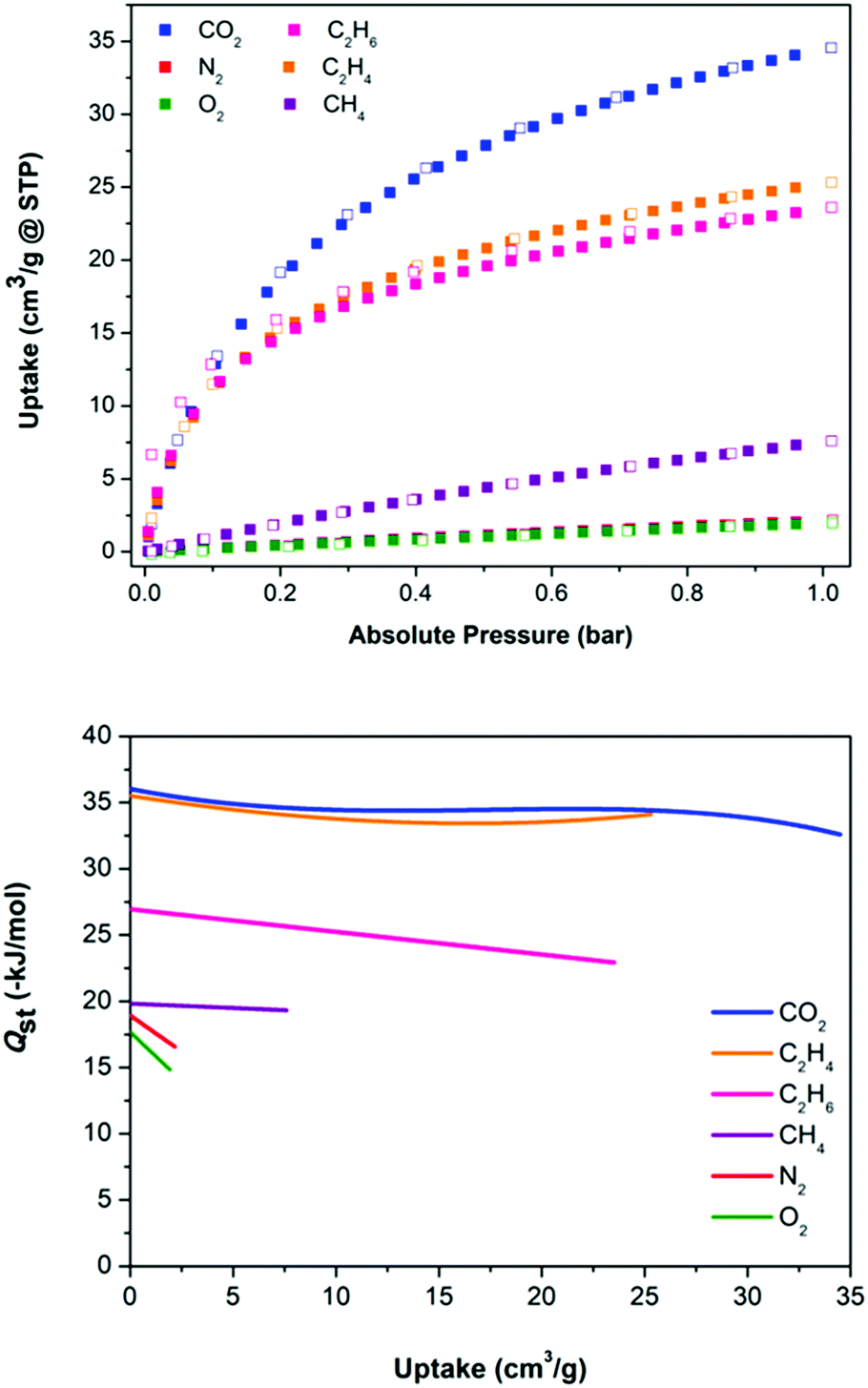 | ||
| Fig. 3 (top) Adsorption isotherms for 1 at 295 K. (bottom) Plot of Qst as a function of loading for sorbates within 1. | ||
| Void spacea | N2 uptakeb (cm3 g−1), Qstc (−kJ mol−1) | CO2 uptakeb (cm3 g−1), Qstc (−kJ mol−1) | CH4 uptakeb (cm3 g−1), Qstc (−kJ mol−1) | C2H4 uptakeb (cm3 g−1), Qstc (−kJ mol−1) | C2H6 uptakeb (cm3 g−1), Qstc (−kJ mol−1) | O2 uptakeb (cm3 g−1), Qstc (−kJ mol−1) |
|---|---|---|---|---|---|---|
| a The accessible void space and micropore volume were calculated with the program PLATON15 using a probe radius of 1.2 Å. b Uptakes reported at 1 bar and 295 K (cm3 g−1). c Sorption data at 273 and 295 K were fit to a virial function from which the isosteric enthalpy of adsorption (Qst, kJ mol−1) was calculated. | ||||||
| 26% | 2.1, 18.9 | 34.5, 36.0 | 7.5, 19.8 | 25.3, 35.5 | 23.6, 26.9 | 1.9, 17.7 |
From the range of adsorbates tested, the uptakes of CO2 and N2 are striking (Table 1). They reveal a significant uptake of CO2 and very little of N2: at 295 K and 1 bar, uptake capacities of CO2 and N2 are equal to 34.5 and 2.1 cm3 g−1, respectively. This behaviour is a highly desirable feature touted for materials used in CO2 removal from flue gas streams and has been observed in other microporous physisorbent materials.16,17 The selectivity for CO2 over N2 in a 1:1 mixture was calculated using IAST to vary between 75 (low pressure) and 40 (1 bar), as shown in Fig. S25 (ESI†). The selectivity of 1 for this gas pair is higher than those reported for a number of well-known MOFs, including CuBTC,18 SIFSIX-1-Cu6b and PCN-61.19 The selectivity for CO2 over methane is also impressive, varying between 16 and 20 (Fig. S25, ESI†). Breakthrough curves for these gas pairs were simulated. These predict that 1 has the clear potential to separate CO2/N2 and CO2/CH4 mixtures on a bulk scale.
To evaluate the strength of interactions between adsorbates and 1, their isosteric heats of adsorption (Qst) were calculated (Fig. 3 and Fig. S11–S23, ESI†). For the six adsorbates, the magnitude of the Qst values follow the same trend of the uptake capacities (Table 1). Notably, for CO2 the enthalpy at low loading is equal to 36 kJ mol−1, and for N2 19 kJ mol−1. These values are on par with expectations based on the quadrupole moments and polarisabilities of these adsorbates, and are comparable with values observed for MOFs and related porous materials.20 Notably, the Qst for N2 is higher than that of O2. This is the reverse of that expected on the basis of van der Waals interactions and is due to the quadrupole moment of N2 and its interaction with the strong electric field gradient of 1 that stems from the presence of the BF4− anions in the channels. A similar situation is seen in zeolites, which are also highly polar. For MOFs the situation is usually the reverse (O2 > N2) since their pores are typically less polar.
In conclusion, a new molecular porous material constructed from dinuclear mesocate complexes [Fe2(L)3](BF4)4, 1, has been detailed and represents a rare example of an extrinsically porous material. The formation of extensive supramolecular interactions between mesocate complexes serve to sustain an overall 3D architecture. 1D channels protruding through the structure have been studied by gas sorption experiments and reveal high CO2 over N2 selectivity, surpassing several well-known MOFs. These results serve to emphasise the advantages offered by MPMs for selective gas sorption and provide design strategies for establishing new variants. Current work in our laboratory is looking at new variants of compound 1 by using related ligand sets in combination with other metal salts and results from these studies will be reported in due course.
The authors gratefully acknowledge the MacDiarmid Institute, the MBIE Catalyst Fund, and the Royal Society of New Zealand Marsden Fund; the Dumont d’Urville NZ-France Science & Technology Support Programme for financial support and the New Zealand France Friendship Fund for the award of an Excellence Scholarship to BHW. The University of Bordeaux, the CNRS, the Région Nouvelle Aquitaine, the MOLSPIN COST action CA15128 and the GdR MCM-2: Magnétisme et Commutation Moléculaires is also thanked for their financial support. BHW gratefully acknowledges the receipt of the Roper Scholarship (UC). We thank Dr Dan Preston (UC) for assistance with Fig. 1.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) F. Rouquerol, J. Roquerol, D. S. W. Sing, P. Llewellyn and G. Maurin, Adsorption by Powders and Porous Solids: Principles, Methodology and Applications, Elsevier, Poland, 2nd edn, 2014 Search PubMed; (b) P. A. Wright, Microporous Framework Solids, Royal Society of Chemistry, Cambridge, 2008 Search PubMed; (c) H. Yang, S. J. Bradley, X. Wu, A. Chan, G. I. N. Waterhouse, T. Nann, J. Zhang, P. E. Kruger, S. Ma and S. G. Telfer, ACS Nano, 2018, 12, 4594 CrossRef CAS PubMed; (d) H. Yang, S. J. Bradley, A. Chan, G. I. N. Waterhouse, T. Nann, P. E. Kruger and S. G. Telfer, J. Am. Chem. Soc., 2016, 138, 11872 CrossRef CAS PubMed.
- R. Xu, W. Pang, J. Yu, Q. Huo and J. Chen, Chemistry of Zeolites and Related Porous Materials: Synthesis and Structure, John Wiley & Sons Ltd, Singapore, 2007 Search PubMed.
- (a) Metal-Organic Framework Materials, ed. C. M. Lukehart and L. R. MacGillivray, John Wiley & Sons Ltd, United Kingdom, 2014 Search PubMed; (b) The Chemistry of Metal-Organic Frameworks: Synthesis, Characterization, and Applications, ed S. Kaskel, Wiley-VCH Verlag GmbH & Co., 2016 Search PubMed; (c) D. C. Young, H. Yang, S. G. Telfer and P. E. Kruger, Inorg. Chem., 2017, 56, 12224 CrossRef CAS PubMed; (d) H. S. Scott, S. Mukherjee, D. R. Turner, M. I. J. Polson, M. J. Zaworotko and P. E. Kruger, CrystEngComm., 2018, 20, 1193 RSC; (e) S. J. Lee, C. Doussot and S. G. Telfer, Cryst. Growth Des., 2017, 17, 3185 CrossRef CAS; (f) K. M. Patil, S. G. Telfer, S. C. Moratti, O. T. Qazvini and L. R. Hanton, CrystEngComm, 2017, 19, 7236 RSC.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 538 RSC.
- (a) N. B. McKeown, J. Mater. Chem., 2010, 20, 10588 RSC; (b) J. Tian, P. K. Thallapally and B. P. McGrail, CrystEngComm, 2012, 14, 1909 RSC; (c) A. G. Slater and A. I. Cooper, Science, 2015, 348, 6238 CrossRef PubMed; (d) C. G. Bezzu, M. Helliwell, J. E. Warren, D. R. Allan and N. B. McKeown, Science, 2010, 327, 1627 CrossRef CAS PubMed.
- (a) H. Kim, Y. Kim, M. Yoon, S. Lim, S. M. Park, G. Seo and K. Kim, J. Am. Chem. Soc., 2010, 132, 12200 CrossRef CAS PubMed; (b) P. S. Nugent, V. L. Rhodus, T. Pham, K. Forrest, L. Wojtas, B. Space and M. J. Zaworotko, J. Am. Chem. Soc., 2013, 135, 10950 CrossRef CAS PubMed.
- T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973 CrossRef CAS PubMed.
- S. Wang, T. Zhao, G. Li, L. Wojtas, Q. Huo, M. Eddaoudi and Y. Liu, J. Am. Chem. Soc., 2010, 132, 18038 CrossRef CAS PubMed.
- (a) A. C. Sudik, A. R. Millward, N. W. Ockwig, A. P. Côté, J. Kim and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 7110 CrossRef CAS PubMed; (b) M. B. Duriska, S. M. Neville, J. Lu, S. S. Iremonger, J. F. Boas, C. J. Kepert and S. R. Batten, Angew. Chem., Int. Ed., 2009, 121, 9081 CrossRef; (c) G. A. Craig, P. Larpent, S. Kusaka, R. Matsuda, S. Kitagawa and S. Furukawa, Chem. Sci., 2018, 9, 6463 RSC; (d) J.-R. Li and H.-C. Zhou, Nat. Chem., 2010, 2, 893 CrossRef CAS PubMed; (e) W. Lu, D. Yuan, A. Yakovenko and H.-C. Zhou, Chem. Commun., 2011, 47, 4968 RSC; (f) J. M. Teo, C. J. Coghlan, J. D. Evans, E. Tsivion, M. Head-Gordon, C. J. Sumby and C. J. Doonan, Chem. Commun., 2016, 52, 276 RSC; (g) K. Xiong, F. Jiang, Y. Gai, D. Yuan, L. Chen, M. Wu, K. Su and M. Hong, Chem. Sci., 2012, 3, 2321 RSC; (h) D. Preston, K. F. White, J. E. M. Lewis, R. A. S. Vasdev, B. F. Abrahams and J. D. Crowley, Chem. – Eur. J., 2017, 23, 10559 CrossRef CAS PubMed.
- H. Wang, B. Li, H. Wu, T.-L. Hu, Z. Yao, W. Zhou, S. Xiang and B. Chen, J. Am. Chem. Soc., 2015, 137, 9963 CrossRef CAS PubMed.
- J. Thomas-Gipson, G. Beobide, O. Castillo, M. Fröba, F. Hoffmann, A. Luque, S. Pérez-Yáñez and P. Román, Cryst. Growth Des., 2014, 14, 4019 CrossRef CAS.
- J. Tian, H. Wang, D.-W. Zhang, Y. Liu and Z.-T. Li, Natl. Sci. Rev., 2017, 4, 426 CrossRef.
- (a) R. J. Archer, C. S. Hawes, G. N. L. Jameson, V. McKee, B. Moubaraki, N. F. Chilton, K. S. Murray, W. Schmitt and P. E. Kruger, Dalton Trans., 2011, 40, 12368 RSC; (b) D. Pelleteret, R. Clérac, C. Mathonière, E. Harté, W. Schmitt and P. E. Kruger, Chem. Commun., 2009, 221 RSC; (c) R. J. Archer, H. S. Scott, M. I. J. Polson, B. E. Williamson, C. Mathonière, M. Rouzières, R. Clérac and P. E. Kruger, Dalton Trans., 2018, 47, 7965 RSC.
- Single crystal X-ray diffraction data were also collected at 298 K. While the quality of the crystal was poor, the data gave a reasonable structural solution in the same P63/m space group. The average Fe–N bond length of 1.958(10) Å reveals that the Fe(II) centre remains in the low spin state at 298 K.
- A. L. Spek, PLATON SQUEEZE: a tool for the calculation of the disordered solvent contribution to the calculated structure factors, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 9 CrossRef CAS PubMed.
- D. Aaron and C. Tsouris, Sep. Sci. Technol., 2005, 40, 321 CrossRef CAS.
- (a) J. Thomas-Gipson, G. Beobide, O. Castillo, J. Cepeda, A. Luque, S. Pérez-Yáñez, A. T. Aguayo and P. Román, CrystEngComm, 2011, 13, 3301 RSC; (b) K. M. Ok, J. Sung, G. Hu, R. M. J. Jacobs and D. O’Hare, J. Am. Chem. Soc., 2008, 130, 3762 CrossRef CAS PubMed; (c) T. K. Maji, R. Matsuda and S. Kitagawa, Nat. Mater., 2007, 6, 142 CrossRef CAS PubMed.
- Z. Zhang, Y. Zhao, Q. Gong, Z. Lib and J. Li, Chem. Commun., 2013, 49, 653 RSC.
- B. Zheng, J. Bai, J. Duan, L. Wojtas and M. J. Zaworotko, J. Am. Chem. Soc., 2011, 133, 748 CrossRef CAS PubMed.
- (a) H. S. Scott, N. Ogiwara, K.-J. Chen, D. G. Madden, T. Pham, K. Forrest, B. Space, S. Horike, J. J. Perry, S. Kitagawa and M. J. Zaworotko, Chem. Sci., 2016, 7, 5470 RSC; (b) K.-J. Chen, H. S. Scott, D. G. Madden, T. Pham, A. Kumar, A. Bajpai, M. Lusi, K. A. Forrest, B. Space, J. J. Perry and M. J. Zaworotko, Chem, 2016, 1, 753 CrossRef CAS; (c) P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details; SC-XRD information; H-bond tables; crystal packing diagrams; TGA; PXRD; sorption data. CCDC 1841640. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc07227b |
| This journal is © The Royal Society of Chemistry 2018 |