
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular electronic spin qubits from a spin-frustrated trinuclear copper complex†
Benjamin
Kintzel‡
a,
Michael
Böhme‡
 a,
Junjie
Liu
b,
Anja
Burkhardt
c,
Jakub
Mrozek
b,
Axel
Buchholz
a,
Arzhang
Ardavan
b and
Winfried
Plass
*a
a,
Junjie
Liu
b,
Anja
Burkhardt
c,
Jakub
Mrozek
b,
Axel
Buchholz
a,
Arzhang
Ardavan
b and
Winfried
Plass
*a
aInstitut für Anorganische und Analytische Chemie, Friedrich-Schiller-Universität Jena, Humboldtstraße 8, 07745 Jena, Germany. E-mail: sekr.plass@uni-jena.de; Fax: +49 3641 948132; Tel: +49 3641 948130
bThe Clarendon Laboratory, Department of Physics, University of Oxford, Oxford OX1 3PU, UK
cPhoton Science, DESY, Notkestrasse 85, 22607 Hamburg, Germany
First published on 2nd October 2018
Abstract
The trinuclear copper(II) complex [Cu3(saltag)(py)6]ClO4 (H5saltag = tris(2-hydroxybenzylidene)triaminoguanidine) was synthesized and characterized by experimental as well as theoretical methods. This complex exhibits a strong antiferromagnetic coupling (J = −298 cm−1) between the copper(II) ions, mediated by the N–N diazine bridges of the tritopic ligand, leading to a spin-frustrated system. This compound shows a T2 coherence time of 340 ns in frozen pyridine solution, which extends to 591 ns by changing the solvent to pyridine-d5. Hence, the presented compound is a promising candidate as a building block for molecular spintronics.
Quantum technology is one of the challenging fields of the current century due to potential major advances in information technology.1 Qubits are the basic building blocks of quantum computers and in a straightforward approach can be realized by nuclear or electronic systems.2 Although nuclear spins generally show longer coherence times than electronic spins, the latter have been shown to be suitable in terms of molecular magnets.3 In fact, electronic spins related to magnetic molecules offer important advantages, as their structural and electronic properties can be tuned by chemical means and are easier to manipulate by employing electric fields.4 Along this line, homotrinuclear complexes of paramagnetic metal ions have been attracting interest for several reasons. For magnetically anisotropic metal ions with large spins this includes cases which have been investigated with regard to their spin interaction in the triangle,5 tuning single-molecule magnets6 and single-molecule toroic behavior.7 In contrast to ferromagnetically coupled molecular triangles,8 an antiferromagnetic coupling causes situations which Kahn described as ranging from “competing spin interactions” to “degenerate frustration”.9 For molecules with an equilateral triangle topology and strict C3 symmetric arrangement, antiferromagnetic interactions lead to specific properties. In the case of ions with integer spin like Ni(II) this results in a diamagnetic ground state (S = 0),10 whereas systems based on non-integer spin such as Cu(II) ions give rise to the magnetic phenomenon of two degenerate spin ground states, the so-called spin frustration.9,11 This situation in molecular systems is predicted to give rise to spin-electric coupling that allows manipulating the molecular spin states.12 For practical implementation a large antiferromagnetic isotropic exchange interaction to provide an energetically well separated chiral spin ground state and small antisymmetric exchange interactions are favorable molecular properties.13 However, this combination is hardly present in most complexes reported so far,14 although such systems are highly demanded for quantum computing and spintronic applications.15
In this work, we present the synthesis and characterization of the trinuclear Cu(II) complex [Cu3(saltag)(py)6]ClO4 (Cu3saltag), which is based on the central tritopic triaminoguanidine derived ligand (H5saltag = tris(2-hydroxybenzylidene)triaminoguanidine). The compound was obtained by a stepwise synthetic approach utilizing three equivalents of Cu(II) perchlorate, one equivalent of the ligand hydrochloride (H5saltag·HCl), six equivalents of NEt3 as a base, and finally an excess of pyridine to saturate the coordination sphere of the copper centers (see the ESI†). Cu3saltag crystallizes as Cu3saltag·1/2([Hpy]ClO4)·H2O in the trigonal space group P31c (Table S1, ESI†). The unit cell contains two crystallographically independent trinuclear monocationic complexes [Cu3(saltag)(py)6]+ (Fig. 1 and Fig. S1, ESI†), one pyridinium cation, three perchlorate anions, and two co-crystallized water molecules. Each complex cation is centered on one of the two distinct crystallographic 3-fold axes, so that within the independent cations all Cu centers are equivalent. The general arrangement of the individual components along the two crystallographic 3-fold axes is illustrated in Fig. S2 (ESI†). The phase purity of the bulk material was confirmed by X-ray powder diffraction (Fig. S3, ESI†).
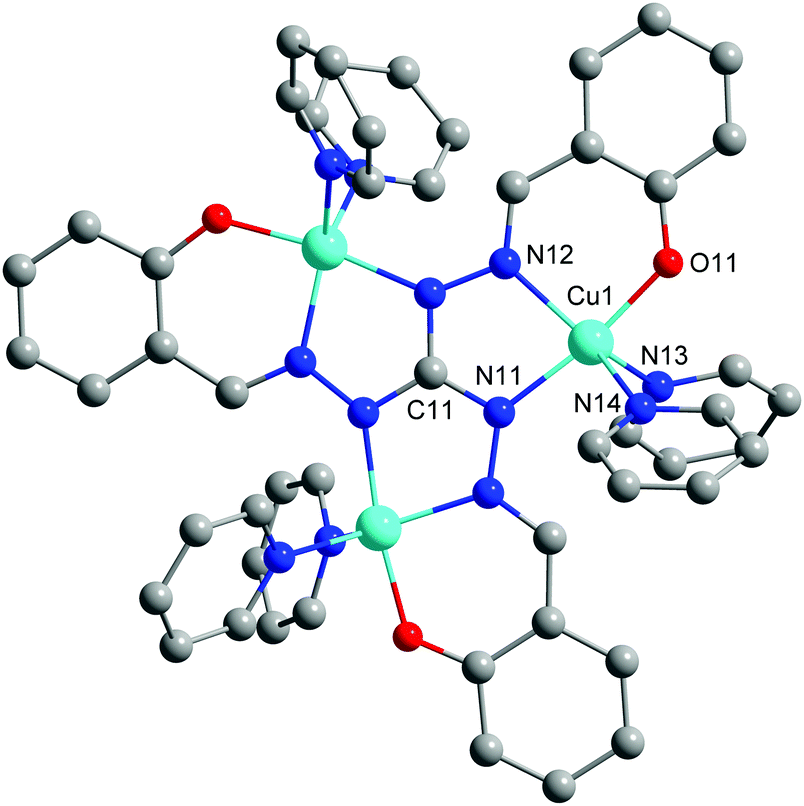 | ||
| Fig. 1 Molecular structure for one of the independent cationic complexes [Cu3(saltag)(py)6]+. Hydrogen atoms are omitted for clarity. | ||
The copper centers of the two complex cations (Cu1 and Cu2) are coordinated in a compressed trigonal bipyramidal fashion by an [N4O] donor set, where an [N2O] pocket is provided by the central saltag5− ligand and two pyridine molecules saturate the remaining coordination sites (Fig. S4, ESI†). For selected bond lengths and angles see Table S2 (ESI†). The donor atoms O11 (O21) and N11 (N21) are at the apical positions of the bipyramid featuring the shortest bond lengths among the coordinative bonds (Cu1–O11: 190.7(3), Cu2–O21: 190.0(2); Cu1–N11: 197.4(3), Cu2–N21: 197.9(3) pm). The Cu–N bonds of the two pyridine ligands in the trigonal plane are rather elongated and vary between 211.9(3) and 218.5(3) pm for both complex molecules. The distorted trigonal bipyramidal coordination environment is confirmed by continuous shape measures, which give the best fit for a trigonal bipyramid (Cu1: S(D3h) = 1.694; Cu2: S(D3h) = 1.705).16
The three perchlorate anions present in the crystal structure, which counterbalance the charge of the monocationic complex molecules and the co-crystallized pyridinium cation, are located with the chlorine atom on special positions on either of the crystallographic 3-fold axes with one of the Cl–O bonds aligned along these axes. For all three perchlorate anions this leads to ClO4⋯Cn1 ionic interactions with the central carbon atom of the triaminoguanidine ligand (Fig. S5 and Table S3, ESI†). In the case of the complex molecule with Cu1 two perchlorate anions are embedded in the two propeller shaped cavities formed by the pyridine co-ligands (Fig. S6, ESI†). On the other hand, for the second complex molecule (Cu2) only one perchlorate anion is present (Fig. S7, ESI†), with the remaining void on the opposite site filled with co-crystallized water molecules (Fig. S2, ESI†). DFT studies for these arrangements reveal remarkably high electrostatic interaction energies with the cationic trinuclear complexes ([Cu3(saltag)(py)6]+⋯ClO4− binding energies: Cu1, 69 and 63; Cu2, 60 kcal mol−1) (Table S4, ESI†). These attractive energies contain a small amount of py⋯ClO4− interaction (Cu1: 17 and 14; Cu2: 13 kcal mol−1; see Table S5 and Fig. S8, ESI†). Interestingly, structural optimization studies with DFT showed that the perchlorate anions are most probably the reason for stabilizing the copper(II) ions in a trigonal bipyramidal coordination sphere in the crystalline material (Fig. S9, ESI†). This is confirmed by the optimized structure obtained without perchlorate anions being present, for which the trinuclear complex exhibits a somewhat more square pyramidal coordination environment (Fig. S9, ESI†).
Additionally, also π–π interactions between the pyridine co-ligands of the crystallographically independent complex molecules are observed (Fig. S5, ESI†). Hence, each trinuclear cationic complex is linked to three neighboring complexes of the other crystallographic type, forming an infinite two-dimensional double layer within the crystallographic ab plane (Fig. S10 and S11, ESI†).
Temperature-dependent magnetic susceptibility measurements for Cu3saltag (Fig. 2) revealed a room temperature χMT value of 0.66 cm3 K mol−1, which is significantly lower than the spin-only value expected for three independent Cu(II) ions with S = 1/2 (1.13 cm3 K mol−1). Upon lowering the temperature, the χMT value further decreases and reaches a plateau at roughly 80 K. This behavior is indicative of a very strong antiferromagnetic intramolecular magnetic exchange between the three copper(II) centers. At temperatures below 10 K, the χMT value again decreases, which indicates the presence of very weak intermolecular interactions. The data were fitted to the spin Hamiltonian given in eqn (1) using the software PHI17 (see the ESI†). The spin coupling scheme in combination with the resulting magnetic states is depicted in Fig. S12 (ESI†). The best fit was obtained with the parameters given in Table S6 (ESI†), revealing a strong antiferromagnetic exchange (J = −297.8(5) cm−1) and weak antiferromagnetic intermolecular interactions (zJ = −0.048(3) cm−1). Moreover, including antisymmetric exchange in the Hamiltonian does not improve the quality of the obtained fit (Table S7 and Fig. S13, ESI†). In fact, intermolecular interactions better describe the low temperature behavior. The upper limit for a possible contribution from antisymmetric exchange can be estimated to be |Gz| < 1 cm−1 (|Gz/J| < 0.003). Although the effective value for Gz is expected to be significantly lower, even the given upper limit is already well below the usually observed range.14,18
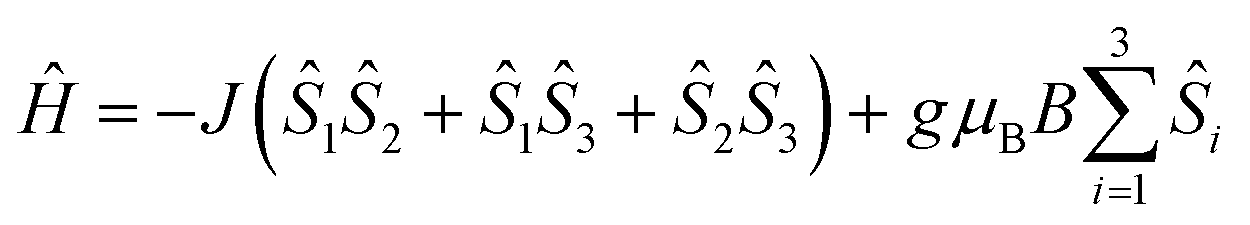 | (1) |
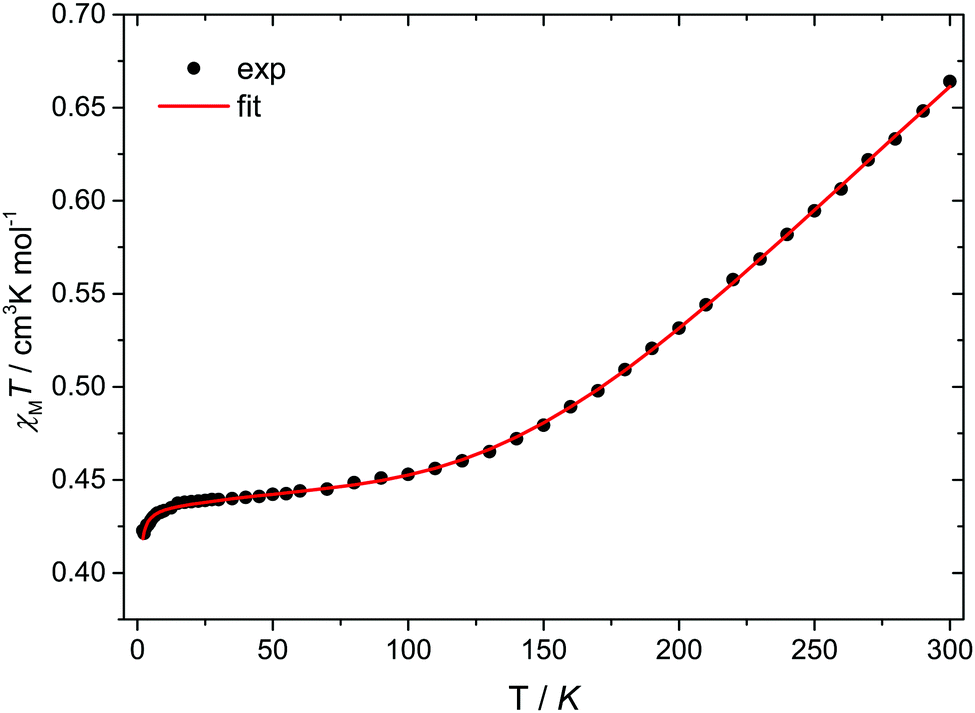 | ||
| Fig. 2 Temperature dependence of χMT for Cu3saltag. The solid red line represents the best fit (see text and Table S6, ESI†). | ||
Broken-symmetry DFT (BS-DFT) calculations were performed to get further insights into the magnetic exchange (Fig. S14, ESI†). The BS-DFT results obtained for pairwise interactions in zinc-replaced model structures verify the strong antiferromagnetic exchange in both complex cations of Cu3saltag (JCu1Cu1 = −281 cm−1; JCu2Cu2 = −274 cm−1; Jav = −278 cm−1; see Table S8, ESI†) and are in good agreement with the experimental value of −297.8(5) cm−1. Alternatively, the exchange coupling can also be directly derived from the energy difference ΔE = EQ − ED = 3/2J, assuming that both the quartet and doublet states are correctly represented by the single determinate solutions for the HS and BS states (ΔE = EHS − EBS), respectively, which, however, slightly underestimates the experimental values (Cu1: −255; Cu2: −249 cm−1; cf. Table S9, ESI†).19 The strong antiferromagnetic exchange within the complex cation is mediated by the N–N diazine bridges, which are formed upon deprotonation of the ligand. Spin-density plots of the high-spin and broken-symmetry states as well as the magnetic orbitals obtained by orbital transformation20 are depicted in Fig. S15–S18 (ESI†). The donor atoms of the [N2O] binding pockets of the saltag5− ligand show a significant spin polarization, whereas the two pyridine nitrogen atoms of the co-ligands are significantly less spin polarized.
For a similar trinuclear complex, [Cu3(saltag)(bpy)3]ClO4 (bpy = 2,2′-bipyridine), also exhibiting molecular C3 symmetry, a slightly stronger antiferromagnetic coupling was observed (J = −324(2) cm−1).21 Since both compounds Cu3saltag and [Cu3(saltag)(bpy)3]ClO4 are based on the same saltag5− ligand, this effect can be attributed to the difference in coordination geometry at the copper centers, which for [Cu3(saltag)(bpy)3]ClO4 is square-pyramidal enforced by the bpy co-ligands and trigonal bipyramidal for Cu3saltag. Moreover, replacing the bpy ligands by pyridine also has a pronounced effect on the crystal packing in Cu3saltag, as it prevents the dimerization of the molecular triangles observed for [Cu3(saltag)(bpy)3]ClO4. For the latter this led to considerable intermolecular exchange interactions at low temperatures (zJ′ = −1.12 cm−1),21 whereas for Cu3saltag the corresponding intermolecular exchange interactions (zJ′ = −0.048 cm−1) are about twenty times smaller.
Continuous wave X-band ESR measurements at 6 K of a powdered crystalline sample of Cu3saltag revealed axial type anisotropy for the exclusively populated magnetic ground state (see Fig. S19 and Table S10, ESI†). A suitable fit for the data is obtained using a g factor with the two principal components g⊥ = 2.04 and g‖ = 2.19 together with a strain parameter for g‖ of 0.20. The latter is indicative of a distribution of individual g‖ values due to minor differences in the coordination environment of the copper(II) ions along their magnetic z axis. This observation appears reasonable based on the presence of two different complex cations in the structure and the rigid ligand plane provided by the saltag5− ligand, the latter being in contrast to the rather mobile monodentate pyridine co-ligands. Similarly, for X-band ESR spectra of frozen solutions of Cu3saltag in pyridine (pyridine-d5) at 3 K (see Fig. S19, ESI†), an axial signal was observed, which can be fitted with a rather similar g⊥ of 2.05 (2.04) and but requires a slightly larger g‖ of 2.29 (2.30) as compared to the solid state spectra. However, also for the spectra of the frozen solutions an identical strain parameter g‖ of 0.20 (0.25) was necessary to reproduce the experimental results in the fit. Nevertheless, upon dissolving Cu3saltag in pyridine (pyridine-d5) we expect the intermolecular π–π as well as the ionic perchlorate interactions to be strongly reduced. Therefore, it is tempting to attribute these changes in interactions to the observed difference in the g‖ values between the bulk material and pyridine solutions. The latter is indicative of a slight variation of the copper(II) coordination environments between both cases.
Ab initio calculations were carried out for both independent copper(II) centers (Cu1 and Cu2) to investigate their magnetic anisotropy (Fig. S20 and Table S11, ESI†). For both centers a 2D multiplet with a 2A′ ground state is obtained as expected for copper(II) in a trigonal bipyramidal coordination sphere (for relative CASSCF and CASPT2 energies see Table S12 (ESI†); for spin–orbit coupled energies see Table S13, ESI†). Since the 2A′ ground state does not show a first-order spin–orbit contribution, any magnetic anisotropy is solely based on the second-order spin–orbit contributions of the excited 2E′ and 2E′′ states. The magnetic anisotropy of both copper(II) centers shows a strong rhombic distortion (gx ≠ gy ≠ gz, see Table S14 and Fig. S21 and S22, ESI†). The hard axis of magnetization gz (Cu1/Cu2: 2.019/2.019) at both metal centers coincides with the Cun–On1 bond vector (angle Cu1/Cu2: 1.3°/1.9°), whereas the intermediate axis gy (Cu1/Cu2: 2.255/2.250) is nearly parallel to the Cun–Nn2 bond vector (angle Cu1/Cu2: 9.0°/10.3°) and the easy axis of magnetization gx is parallel to the molecular C3 axis (Cu1/Cu2: 2.413/2.413). Utilizing the single-ion anisotropies to simulate the magnetic susceptibility of the trinuclear cationic complexes (see the ESI†) shows good agreement with the experiment (see Fig. S23, ESI†), but slightly too large χMT values. Such a shift in χMT values is based on an overestimation in the corresponding g factors and is a known phenomenon.22 The calculated g factors for the molecular framework are given in Table S15 (ESI†). In fact, the situation becomes somewhat different when going from the single-ion anisotropy to the anisotropy of the trinuclear framework. For the latter, an axial system (Cu1: g⊥ = 2.158 and g‖ = 2.425; Cu2: g⊥ = 2.158 and g‖ = 2.423) is confirmed, where the g‖ axis coincides with the C3 axis of the molecular framework.
For perspective applications, spin relaxation times are a crucial feature of the system. They can usually be enhanced via spatial separation of the spin centers.23 Therefore, we performed spin-relaxation time measurements via echo-detected X-band ESR experiments in frozen pyridine and pyridine-d5 solutions of Cu3saltag (see the ESI†). The data were fitted according to eqn (2) to determine the T2 coherence times depending on the used solvent. In pyridine a T2 time of 340 ns was obtained (see Fig. S24, ESI†), whereas in pyridine-d5 this coherence time is significantly longer (591 ns; see Fig. 3). Fitting the echo decay gives a stretch parameter of x = 0.73, suggesting that the coherence time is limited by spectral diffusion.24 This could be due to ligand rotation and libration processes related to the flexible pyridine ligands.25 Such processes could possibly be suppressed by replacing them with sterically demanding ligands, further increasing the coherence time.
Y(t) = Y(0)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−(T2/2τ)x) exp(−(T2/2τ)x) | (2) |
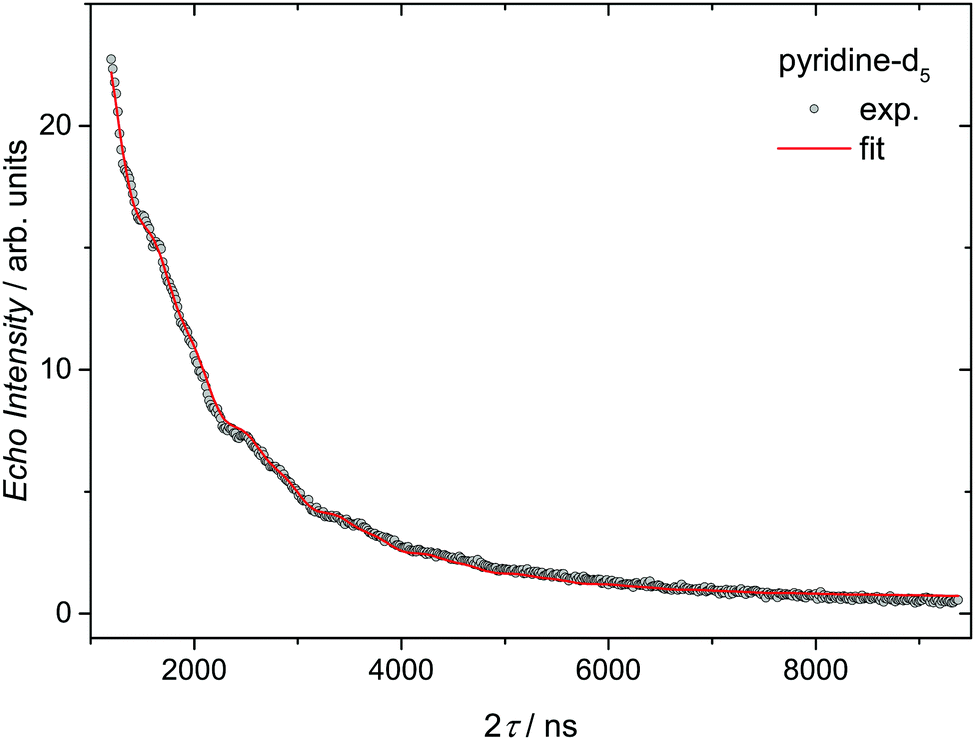 | ||
| Fig. 3 Decay of the Hahn-echo intensity for Cu3saltag in a frozen pyridine-d5 solution measured at 3 K. The solid red line shows the best fit according to eqn (2). The fit assumes that the oscillation in the signal is due to the ESEEM effect dominated by a single harmonic at the deuterium Zeeman frequency.7 | ||
In conclusion, the spin-frustrated trinuclear copper(II) complex Cu3saltag with large antiferromagnetic exchange was synthesized. The magnetic properties were characterized by experimental and theoretical methods. Contributions from antisymmetric exchange are estimated to be well below |Gz/J| < 0.003, making the system favorable for future practical implementation. Moreover, the compound shows a T2 coherence time of 591 ns in a frozen pyridine-d5 solution. These findings make Cu3saltag a promising candidate for further investigations of its applicability as a qubit and of spin-electric couplings.
We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at beamline P11, PETRA III. The authors would like to acknowledge networking support by the MolSpin COST Action CA15128. JM would like to thank EPSRC and Magdalen College, Oxford, for a doctoral studentship.
Conflicts of interest
There are no conflicts to declare.References
- (a) M. Le Bellac, Quantum Computation and Quantum Information, Cambridge University Press, Cambridge, 2006 Search PubMed; (b) M. A. Nielsen and I. L. Chuang, Quantum Computation and Quantum Information, Cambridge University Press, Cambridge, 2010 CrossRef.
- (a) K. Bader, D. Dengler, S. Lenz, B. Endeward, S.-D. Jiang, P. Neugebauer and J. van Slageren, Nat. Commun., 2014, 5, 5304 CrossRef CAS PubMed; (b) J. M. Zadrozny, J. Niklas, O. G. Poluektov and D. E. Freedman, ACS Cent. Sci., 2015, 1, 488–492 CrossRef CAS PubMed; (c) M. Atzori, L. Tesi, E. Morra, M. Chiesa, L. Sorace and R. Sessoli, J. Am. Chem. Soc., 2016, 138, 2154–2157 CrossRef CAS PubMed; (d) M. J. Graham, J. M. Zadrozny, M. S. Fataftah and D. E. Freedman, Chem. Mater., 2017, 29, 1885–1897 CrossRef CAS.
- A. Ardavan, O. Rival, J. J. Morton, S. J. Blundell, A. M. Tyryshkin, G. A. Timco and R. E. Winpenny, Phys. Rev. Lett., 2007, 98, 057201 CrossRef PubMed.
- (a) F. Troiani and M. Affronte, Chem. Soc. Rev., 2011, 40, 3119–3129 RSC; (b) J. Lehmann, A. Gaita-Ariño, E. Coronado and D. Loss, J. Mater. Chem., 2009, 19, 1672–1677 RSC.
- D. Plaul, M. Böhme, S. Ostrovsky, Z. Tomkowicz, H. Görls, W. Haase and W. Plass, Inorg. Chem., 2018, 57, 106–119 CrossRef CAS PubMed.
- (a) D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, Oxford, 2006 CrossRef; (b) L. Gregoli, C. Danieli, A.-L. Barra, P. Neugebauer, G. Pellegrino, G. Poneti, R. Sessoli and A. Cornia, Chem. – Eur. J., 2009, 15, 6456–6467 CrossRef CAS PubMed; (c) T. Glaser, M. Heidemeier and R. Fröhlich, C. R. Chim, 2007, 10, 71–78 CrossRef CAS.
- (a) L. Ungur, S.-Y. Lin, J. Tang and L. F. Chibotaru, Chem. Soc. Rev., 2014, 43, 6894–6905 RSC; (b) J. Luzon, K. Bernot, I. J. Hewitt, C. E. Anson, A. K. Powell and R. Sessoli, Phys. Rev. Lett., 2008, 100, 247205 CrossRef PubMed; (c) L. Chibotaru, L. Ungur and A. Soncini, Angew. Chem., 2008, 120, 4194–4197 CrossRef.
- E. T. Spielberg, M. Fittipaldi, D. Geibig, D. Gatteschi and W. Plass, Inorg. Chim. Acta, 2010, 363, 4269–4276 CrossRef CAS.
- O. Kahn, Chem. Phys. Lett., 1997, 265, 109–114 CrossRef CAS.
- (a) A. E. Ion, E. T. Spielberg, H. Görls and W. Plass, Inorg. Chim. Acta, 2007, 360, 3925–3931 CrossRef CAS; (b) T. Matsumoto, Y. Sato, T. Shiga and H. Oshio, Inorg. Chem. Front., 2015, 2, 725–730 RSC.
- J. Schnack, Dalton Trans., 2010, 39, 4677–4686 RSC.
- (a) M. Trif, F. Troiani, D. Stepanenko and D. Loss, Phys. Rev. Lett., 2008, 101, 217201 CrossRef PubMed; (b) M. Trif, F. Troiani, D. Stepanenko and D. Loss, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 045429 CrossRef; (c) M. F. Islam, J. F. Nossa, C. M. Canali and M. Pederson, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 155446 CrossRef.
- D. Stepanenko, M. Trif, O. Tsyplyatyev and D. Loss, Semicond. Sci. Technol., 2016, 31, 094003 CrossRef.
- S. Ferrer, F. Lloret, E. Pardo, J. M. Clemente-Juan, M. Liu-González and S. García-Granda, Inorg. Chem., 2012, 51, 985–1001 CrossRef CAS PubMed.
- S. Carretta, P. Santini, G. Amoretti, F. Troiani and M. Affronte, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 024408 CrossRef.
- (a) H. Zabrodsky, S. Peleg and D. Avnir, J. Am. Chem. Soc., 1993, 115, 8278–8289 CrossRef CAS; (b) H. Zabrodsky, S. Peleg and D. Avnir, IEEE Trans. Pattern Anal. Mach. Intell., 1995, 17, 1154–1166 CrossRef; (c) M. Pinsky and D. Avnir, Inorg. Chem., 1998, 37, 5575–5582 CrossRef CAS PubMed.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
- M. I. Belinsky, Inorg. Chem., 2008, 47, 3521–3531 CrossRef CAS PubMed.
- (a) J. P. Perdew, A. Savin and K. Burke, Phys. Rev. A: At., Mol., Opt. Phys., 1995, 51, 4531–4541 CrossRef CAS; (b) J. P. Perdew, A. Savin and K. Burke, Phys. Rev. A: At., Mol., Opt. Phys., 1997, 61, 197–205 CAS; (c) E. Ruiz, S. Alvarez, J. Cano and V. Polo, J. Chem. Phys., 2005, 123, 164110 CrossRef PubMed.
- F. Neese, J. Phys. Chem. Solids, 2004, 65, 781–785 CrossRef CAS.
- E. T. Spielberg, A. Gilb, D. Plaul, D. Geibig, D. Hornig, D. Schuch, A. Buchholz, A. Ardavan and W. Plass, Inorg. Chem., 2015, 54, 3432–3438 CrossRef CAS PubMed.
- (a) M. Atanasov, J. M. Zadrozny, J. R. Long and F. Neese, Chem. Sci., 2013, 4, 139–156 RSC; (b) M. Böhme, S. Ziegenbalg, A. Aliabadi, A. Schnegg, H. Görls and W. Plass, Dalton Trans., 2018, 47, 10861–10873 RSC.
- A. Schweiger and G. Jeschke, Principles of pulse electron paramagnetic resonance, Oxford University Press, New York, 2001 Search PubMed.
- S. S. Eaton and G. R. Eaton, in Relaxation Times of Organic Radicals and Transition Metal Ions, ed. L. J. Berliner, G. R. Eaton and S. S. Eaton, Kluwer Academic/Plenum Publishers, New York, 2000, vol. 19, pp. 29–154 Search PubMed.
- C. Wedge, Phys. Rev. Lett., 2012, 108, 107204 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, crystallographic details, computational details, magnetic data, BS-DFT results, ESR data, and ab initio results. CCDC 1862694. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc06741d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |