
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A facile route to 1H- and 2H-indazoles from readily accessible acyl hydrazides by exploiting a novel aryne-based molecular rearrangement†
André
Shamsabadi
 and
Vijay
Chudasama
*
and
Vijay
Chudasama
*
Department of Chemistry, University College London, London, UK. E-mail: v.chudasama@ucl.ac.uk
First published on 11th September 2018
Abstract
Herein we report the transformation of readily synthesised acyl hydrazides into 2-hydrazobenzophenones via a novel molecular rearrangement pathway using aryne chemistry. The developed reaction protocol is performed under relatively mild conditions and is tolerant of a wide variety of functional groups, and the 2-hydrazobenzophenone products provide access to both 1H- and 2H-indazoles from a single intermediate.
The indazole moiety is of great medicinal importance1 and compounds containing the indazole nucleus have recently sparked great interest for use as anti-inflammatory,2 antitumour3,4 and anti-HIV5 agents, and as inhibitors of protein kinase,6 HIV-protease,7 monoamine oxidase8 and N-myristoyltransferase,9 as well as finding use as a biological probe,10 amongst other applications. However, most strategies for the synthesis of indazoles are generally limited by their requirement of using complex multi-step syntheses,11 and/or using harsh reaction conditions that often promote alternative reaction pathways (e.g. undesirable Wolff–Kishner reduction is often observed in hydrazine-based synthesis for the preparation of indazoles)12 and/or poor functional group tolerance.13 This highlights a general need for synthetic routes that facilitate the synthesis of indazoles, especially those that start from readily accessible starting materials.
Over the past few decades, owing in large part to the plethora of new methodologies that have enabled facile access to arynes,14,15 the employment of aryne chemistry in synthesis has seen a major resurgence.16–21 A salient feature of aryne chemistry is the possibility for the formation of C–C and C–heteroatom bonds on aryl rings, resulting in the synthesis of various di- and even tri-substituted arenes (i.e. if the aryne was already substituted).22–24 Owing to this, aryne chemistry has emerged as a very useful method for the synthesis of benzo-fused heterocycles.25 Of particular relevance to this article is the use of aryne chemistry to synthesise indazoles, i.e. through the use of N-tosylhydrazones26 and in situ generation of nitrile imines27 (for 1H-indazoles) or by using sydnones28 (for 2H-indazoles). Whilst these methods have been successfully utilised to selectively afford 1H- or 2H-indazoles, the requirement of a phase transfer catalyst additive, limited scope (i.e. for the synthesis of 1,3-diarylindazoles only), and the reliance on the use of precursors that require lengthy/cumbersome syntheses (respectively), highlights several key limitations. Moreover, none of these syntheses provide flexible access to both 1H- and 2H-indazoles from a single branch point. As such, despite the inherently favourable nature of aryne chemistry to provide benzo-fused heterocycles, there is still significant scope for development in this area in terms of providing routes to indazoles. This is especially pertinent as currently there is no leading synthetic strategy to synthesise both 1H- and 2H-indazoles in a more general sense, i.e. outside the scope of aryne-based chemistry.
Recently our group29,30 and others31–34 have reported on acyl hydrazides as synthetically versatile scaffolds for the synthesis of a wide range of species. More specifically, acyl hydrazides have been employed in the creation of moieties such as amides,35 esters,35 thioesters,35 ketones,36N-acyl carbamates37 and 1,3,4-oxadiazoles,38 as well as being used as building blocks for the creation of bioactive molecules such as hydroxamic acids39 and macrocyclic enamides.40 Approaches to utilise acyl hydrazides in synthesis have largely focused on substituting the hydrazide moiety35,36,39 or alkylating at the relatively acidic N–H position (see Scheme 1).37,40 In this work, it was envisioned that the reaction of an aryne with an acyl hydrazide would exploit both of these synthetic properties to facilitate a molecular rearrangement reaction pathway that would lead to the synthesis of 2-hydrazobenzophenones. These entities could then act as a single branch point for conversion into 1H- or 2H-indazoles by exploiting the removal of the carbamate groups pre- and post-alkylation (Scheme 1).
 | ||
| Scheme 1 General method for the synthesis of 2-hydrazobenzophenones from acyl hydrazides via benzyne chemistry and the proposed mechanism for use as a single branch point for the formation of 1H- and 2H-indazoles. | ||
Our study began with the reaction of acyl hydrazide 1a with benzyne precursor 2 in the presence of a fluoride source tetrabutylammonium difluorotriphenylsilicate (TBAT), with the aim of forming 2-hydrazobenzophenone 3avia a novel molecular rearrangement pathway (see Scheme 4 below for the proposed mechanism). Initially, the reaction was carried out under the conditions developed by Pintori et al. for the aryl insertion of arynes into amide C(O)–N bonds,41i.e. the use of 1.5 equivalents of the benzyne precursor, 2 equivalents of TBAT and toluene as the reaction solvent at 50 °C. To our delight, the reaction proceeded efficiently, resulting in the formation of the desired product 3a in 86% yield (Table 1, entry 1). Lowering the equivalents of the fluoride source in the reaction resulted in incomplete conversion of acyl hydrazide 1a and thus had a negative impact on the yield (Table 1, entries 2 and 3). Whilst other carbamate groups could be used for this transformation (see the ESI† for details), i-Pr carbamates provided the highest yield.
|
|
|||||
|---|---|---|---|---|---|
| Entry | 2 (eq.) | TBAT (eq.) | Base/eq. | Conversion of 1a (%) | 3a (%) |
| Reaction conditions: acyl hydrazide 1a (0.5 mmol, 1 eq.), benzyne precursor 2 (0.75 mmol, 1.5 eq.) and TBAT in toluene (6 mL); 50 °C for 16 h. | |||||
| 1 | 1.5 | 2 | 3 | 100 | 86 |
| 2 | 1.5 | 1.5 | 3 | 76 | 59 |
| 3 | 1.5 | 1 | 5 | 54 | 38 |

With the optimised conditions for the transformation of acyl hydrazides into 2-hydrazobenzophenones in hand, we took the opportunity to investigate the applicability of our protocol for the formation of various 2-hydrazobenzophenones (Scheme 2). A range of acyl hydrazides were examined (1a–1l) under the developed reaction conditions. All the starting acyl hydrazides were prepared in good yields using our previously reported procedure for the hydroacylation of azodicarboxylates (see the ESI† for details).29,30
 | ||
| Scheme 2 Scope of reaction with respect to acyl hydrazide 1. Reaction conditions: acyl hydrazides 1a–1l (0.5 mmol, 1 eq.), benzyne precursor 2 (0.75 mmol, 1.5 eq.) and TBAT (1 mmol, 2 eq.) in toluene (6 mL); 50 °C for 16 h. | ||
To our delight, the reaction of acyl hydrazides 1a–1l with benzyne was tolerant of various functional groups on the aromatic acyl hydrazide motif, e.g. electron-withdrawing (halo, trifluoromethyl, ester, and nitro), electron-rich (methyl and methoxy) and electron-neutral (hydrogen) functionalities (Scheme 2). Unfortunately, the presence of ortho-functional groups on the aryl ring resulted in a lower yield of the desired products (3k and 3l). This is likely due to increased steric hindrance about the amide-like carbonyl promoting carbanionic attack on a carbamate carbonyl. This hypothesis is further substantiated by the ortho-fluoro variant (with fluoride being a steric isostere for H) affording product 3j in a higher yield, 70%.
To appraise the notion that the cleavage of the carbamate moieties in 2-hydrazobenzophenones would result in subsequent intramolecular dehydrative cyclisation to form desired 1H-indazoles, removal of the carbamates of compound 3a was attempted under a variety of conditions (see Table 2). The use of Lewis acid AlCl3 resulted in incomplete conversion of the starting material 3a and only carbamate 1H-indazole 5 was observed in 30% yield (Table 2, entry 1). Refluxing in AcOH or HCl (3 M) resulted in complete conversion of the starting material and gave access to carbamate 1H-indazole 5 only, in 65% and 71% yields, respectively. Pleasingly, however, the use of either 12 M HCl or KOH in dimethylacetamide resulted in excellent yields, >90%, of the desired 1H-indazole 4a (Table 2, entries 4 and 5).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Reagent | Solvent | Temperature (°C) | Yield 4a (%) | Yield 5 (%) |
| Reaction conditions: 2-hydrazobenzophenone 3a (0.5 mmol) under varying conditions for 16 h. DMA = dimethylacetamide.a 45% conversion of 3a. | |||||
| 1 | AlCl3 | MeNO2 | 60 | 0 | 30a |
| 2 | AcOH | — | Reflux | 0 | 71 |
| 3 | HCl (3 M) | — | Reflux | 0 | 65 |
| 4 | HCl (12 M) | — | Reflux | 91 | 0 |
| 5 | KOH (aq) | DMA | 60 | 92 | 0 |

Having established suitable reaction conditions for the removal of the isopropyl carbamate groups and dehydrative cyclisation, the previously formed library of 2-hydrazobenzophenones 3a–3l were subjected to the relatively mild basic reaction conditions for the formation of various 1H-indazoles (Scheme 3). Most pleasingly, excellent yields were observed across the series with the exception of the 2-hydrazobenzophenone containing the nitro functionality. Fortunately, and highlighting the advantage of being able to employ either basic of acidic conditions, subjecting nitro 2-hydrazobenzophenone 3i to the developed acidic reaction conditions resulted in a high yield, 79%. We also note that 1H-indazoles can be readily and selectively 1H-alkylated using one of several reported procedures.42–44
 | ||
Scheme 3 Scope of reaction with respect to acyl hydrazide 3. Reaction conditions: 2-hydrazobenzophenones 3a–3l (0.25 mmol, 1 eq.) and KOH (1.00 mmol, 4 eq. in H2O (3 mL)) in DMA (5 mL); 60 °C for 16 h. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) Reaction was carried out at 100 °C. bUsing 3e as acyl hydrazide. cHCl (12 M) conditions. Reaction was carried out at 100 °C. bUsing 3e as acyl hydrazide. cHCl (12 M) conditions. | ||
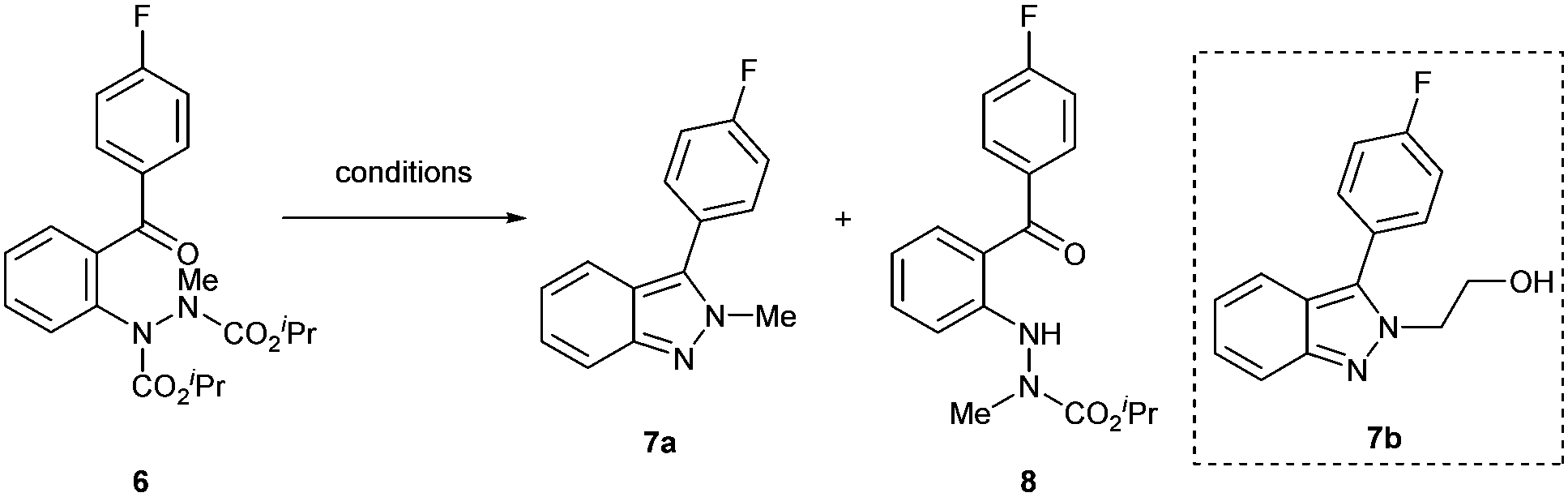
Compared to 1H-indazoles, 2H-indazoles have historically been more challenging to prepare.45 As a result, 2H-indazoles have been far less studied. From the outset, we wanted to establish a method in which the synthesis of N-substituted 2H-indazoles could be enabled from a common intermediate to that used in the synthesis of the 1H-indazoles. We postulated that alkylation of 2-hydrazobenzophenones prior to carbamate removal (and subsequent dehydrative cyclisation) would result in the selective formation of N-substituted 2H-indazoles. To appraise this, alkylation of 2-hydrazobenzophenone 3a was carried out to form alkylated 2-hydrazobenzophenone 6. Compound 6 was then subjected to the various deprotection conditions (see Table 3) to try and form N-methylated 2H-indazole 7a. Fortunately, refluxing HCl (12 M) resulted in the formation of the desired N-methyl 2H-indazole. The use of refluxing AcOH or aqueous KOH in dimethylacetamide only resulted in the cleavage of the internal carbamate, forming the undesired product 8; although we do note that this product was formed selectively and in good yield. Having successfully formed 2H-indazole 7avia alkylation and subsequent removal of carbamates, the reaction sequence was then applied successfully for the formation of functional 2H-indazole 7b (Table 3, inset) in 79% yield from 2-hydrazobenzophenone 3a over 2 steps.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | Reagent | Solvent | Temperature (°C) | Yield 7a (%) | Yield 8 (%) |
| Reaction conditions: alkylated 2-hydrazobenzophenone 6 (0.5 mmol) under varying conditions for 16 h. DMA = dimethylacetamide. | ||||||
| 1 | Me | AcOH | — | Reflux | 0 | 70 |
| 2 | Me | HCl (12 M) | — | Reflux | 91 | 0 |
| 3 | Me | KOH(aq) | DMA | 60 | 0 | 84 |
We next chose to probe the mechanism for the reaction of acyl hydrazide and benzyne. It was important to note, as the starting point, that acyl hydrazides have previously been shown to act as acyl halide analogues,35 and acyl halides and amides have been shown to react with benzynes via an aryl insertion mechanism where the aryne is inserted into the C-halogen σ-bond.41,46 Therefore, aryne insertion into the C(O)–α-nitrogen bond was considered as a plausible mechanistic pathway (Scheme 4, path A). Alternatively, we needed to consider that acyl hydrazides can react nucleophilically at the β-nitrogen atom and that this position may or may not be deprotonated under the reaction conditions (Scheme 4, paths B and C). Therefore, it was necessary to establish whether arynes insert into the C–N σ-bond in acyl hydrazides (Scheme 4, path A) or whether they undergo a novel molecular rearrangement following nucleophilic attack from the more nucleophilic nitrogen atom (Scheme 4, paths B and C).
 | ||
| Scheme 4 Possible reaction mechanisms. | ||
To appraise the feasibility of the reaction pathways, methylated acyl hydrazide 9 was synthesised and subjected to the aryne functionalisation reaction conditions (Scheme 5). Under the reaction conditions, no reaction between the methylated acyl hydrazide 9 and benzyne was observed. If the reaction pathway proceeded via path A, then it would have been expected that the methylated-hydrazobenzophenone product 6 would have formed under these conditions. The almost quantitative recovery of the starting material suggested that the presence of the N–H bond in the starting material is important for the reaction to proceed. Therefore, we next evaluated whether the acyl hydrazide was deprotonated under the reaction conditions, owing to the acidity of the N–H bond. To do this, acyl hydrazide 1a was reacted with 1.1 eq. of iodomethane in the presence of 1.1 eq. of TBAT in toluene at 50 °C (Scheme 5). A control experiment in the absence of TBAT was conducted in parallel. It was observed that in the presence of TBAT, a significant amount of methylated acyl hydrazide 9, 90%, was observed (Scheme 5). In the absence of TBAT, a near quantitative amount of the starting material was recovered. We therefore felt that it was appropriate to conclude that an acyl hydrazide would be significantly deprotonated under the reaction conditions, and suggest that the reaction proceeds through the novel molecular reaction pathway C (Scheme 4).
 | ||
| Scheme 5 Mechanistic studies. | ||
In conclusion, we have exploited a novel molecular rearrangement pathway involving the reaction of readily accessible acyl hydrazides with arynes to provide an intermediate that can be readily converted into either a 1H- or a 2H-indazole, as desired. The developed reaction conditions enable their synthesis from a single intermediate branch point (i.e. providing a highly diverse synthetic route), unveil a novel molecular rearrangement pathway, and tolerate a range of functional groups, and the 1H-indazoles can be formed under either basic or acidic conditions (i.e. a highly flexible route). In view of this, we believe that the protocols which we hereby report for the facile synthesis of 1H- and 2H-indazoles from acyl hydrazides will have wide ranging applications, especially as indazoles are applied in a broad range of medicinal/biological applications. Moreover, the novel reaction pathway we disclose may lead to further exploitation of, or inspiration in, aryne-based molecular rearrangements.
Conflicts of interest
There are no conflicts to declare.Notes and references
- I. Denya, S. F. Malan and J. Joubert, Expert Opin. Ther. Pat., 2018, 28, 441–453 CrossRef PubMed; S. El Kazzouli and G. Guillaumet, Tetrahedron, 2016, 72, 6711–6727 CrossRef; N. A. S. Ali, B. A. Dar, V. Pradhan and M. Farooqui, Mini-Rev. Med. Chem., 2013, 13, 1792–1800 CrossRef PubMed; D. D. Gaikwad, A. D. Chapolikar, C. G. Devkate, K. D. Warad, A. P. Tayade, R. P. Pawar and A. J. Domb, Eur. J. Med. Chem., 2015, 90, 707–731 CrossRef PubMed.
- A. Guglielmotti, A. C. de Joannon, N. Cazzolla, M. Marchetti, L. Soldo, G. Cavallo and M. Pinza, Pharmacol. Res., 1995, 32, 369–373 CrossRef PubMed.
- M. J. Clarke, Coord. Chem. Rev., 2003, 236, 209–233 CrossRef.
- A. Shrivastava, A. K. Chakraborty, N. Upmanyu and A. Singh, Austin J. Anal. Pharm. Chem., 2016, 3, 1–23 Search PubMed.
- J.-H. Sun, C. A. Teleha, J.-S. Yan, J. D. Rodgers and D. A. Nugiel, J. Org. Chem., 1997, 62, 5627–5629 CrossRef.
- K. W. Woods, et al. , Bioorg. Med. Chem., 2006, 14, 6832–6846 CrossRef PubMed.
- M. Patel, J. D. Rodgers, R. J. McHugh, B. L. Johnson, B. C. Cordova, R. M. Klabe, L. T. Bacheler, S. Erickson-Viitanen and S. S. Ko, Bioorg. Med. Chem. Lett., 1999, 9, 3217–3220 CrossRef PubMed.
- N. T. Tzvetkov, S. Hinz, P. Küppers, M. Gastreich and C. E. Müller, J. Med. Chem., 2014, 57, 6679–6703 CrossRef PubMed.
- A. Mousnier, et al. , Nat. Chem., 2018, 10, 599–606 CrossRef PubMed.
- E. Robinson, E. Knight, N. Smoktunowicz, R. C. Chambers, G. G. Inglis, V. Chudasama and S. Caddick, Org. Biomol. Chem., 2016, 14, 3198–3201 RSC.
- X. Xiong, Y. Jiang and D. Ma, Org. Lett., 2012, 14, 2552–2555 CrossRef PubMed.
- K. Lukin, M. C. Hsu, D. Fernando and M. R. Leanna, J. Org. Chem., 2006, 71, 8166–8172 CrossRef PubMed.
- C. S. Cho, D. K. Lim, N. H. Heo, T.-J. Kim and S. C. Shim, Chem. Commun., 2004, 104–105 RSC.
- Y. Himeshima, T. Sonoda and H. Kobayashi, Chem. Lett., 1983, 1211–1214 CrossRef.
- F. I. M. Idiris and C. R. Jones, Org. Biomol. Chem., 2017, 15, 9044–9056 RSC.
- P. M. Tadross and B. M. Stoltz, Chem. Rev., 2012, 112, 3550–3577 CrossRef PubMed.
- J. Shi, Y. Li and Y. Li, Chem. Soc. Rev., 2017, 46, 1707–1719 RSC.
- R. W. Hoffmann and K. Suzuki, Angew. Chem., Int. Ed., 2013, 52, 2655–2656 CrossRef PubMed.
- J.-A. García-López and M. F. Greaney, Chem. Soc. Rev., 2016, 45, 6766–6798 RSC.
- S. S. Bhojgude, A. Bhunia and A. T. Biju, Acc. Chem. Res., 2016, 49, 1658–1670 CrossRef PubMed.
- C. Holden and M. F. Greaney, Angew. Chem., Int. Ed., 2014, 53, 5746–5749 CrossRef PubMed.
- F. I. M. Idiris, C. E. Majesté, G. B. Craven and C. R. Jones, Chem. Sci., 2018, 9, 2873–2878 RSC.
- L.-N. Guo, H. Wang and X.-H. Duan, Org. Biomol. Chem., 2016, 14, 7380–7391 RSC.
- J. Shi, D. Qiu, J. Wang, H. Xu and Y. Li, J. Am. Chem. Soc., 2015, 137, 5670–5673 CrossRef PubMed.
- A. V. Dubrovskiy, N. A. Markina and R. C. Larock, Org. Biomol. Chem., 2013, 11, 191–218 RSC.
- P. Li, J. Zhao, C. Wu, R. C. Larock and F. Shi, Org. Lett., 2011, 13, 3340–3343 CrossRef PubMed.
- C. Spiteri, S. Keeling and J. E. Moses, Org. Lett., 2010, 12, 3368–3371 CrossRef PubMed.
- P.-F. Wang, Y.-S. Feng, Z.-F. Cheng, Q.-M. Wu, G.-Y. Wang, L.-L. Liu, J.-J. Dai, J. Xu and H.-J. Xu, J. Org. Chem., 2015, 80, 9314–9320 CrossRef PubMed.
- V. Chudasama, J. M. Ahern, D. V. Dhokia, R. J. Fitzmaurice and S. Caddick, Chem. Commun., 2011, 47, 3269–3271 RSC.
- V. Chudasama, A. R. Akhbar, K. A. Bahou, R. J. Fitzmaurice and S. Caddick, Org. Biomol. Chem., 2013, 11, 7301–7317 RSC.
- G. N. Papadopoulos, D. Limnios and C. G. Kokotos, Chem. – Eur. J., 2014, 20, 13811–13814 CrossRef PubMed.
- I. Ryu, A. Tani, T. Fukuyama, D. Ravelli, S. Montanaro and M. Fagnoni, Org. Lett., 2013, 15, 2554–2557 CrossRef PubMed.
- A. Mariappan, K. Rajaguru, S. Muthusubramanian and N. Bhuvanesh, Tetrahedron Lett., 2015, 56, 338–341 CrossRef.
- A. Shamsabadi and V. Chudasama, Org. Biomol. Chem., 2017, 15, 17–33 RSC.
- A. Maruani, M. T. W. Lee, G. Watkins, A. R. Akhbar, H. Baggs, A. Shamsabadi, D. A. Richards and V. Chudasama, RSC Adv., 2016, 6, 3372–3376 RSC.
- A. R. Akhbar, V. Chudasama, R. J. Fitzmaurice, L. Powell and S. Caddick, Chem. Commun., 2014, 50, 743–746 RSC.
- A. Shamsabadi, J. Ren and V. Chudasama, RSC Adv., 2017, 7, 27608–27611 RSC.
- O. Sugimoto, T. Arakaki, H. Kamio and K. Tanji, Chem. Commun., 2014, 50, 7314 RSC.
- G. N. Papadopoulos and C. G. Kokotos, Chem. – Eur. J., 2016, 22, 6964–6967 CrossRef PubMed.
- Y. J. Kim and D. Lee, Org. Lett., 2004, 6, 4351–4353 CrossRef PubMed.
- D. G. Pintori and M. F. Greaney, Org. Lett., 2010, 12, 168–171 CrossRef PubMed.
- D. J. Slade, N. F. Pelz, W. Bodnar, J. W. Lampe and P. S. Watson, J. Org. Chem., 2009, 74, 6331–6334 CrossRef PubMed.
- A. Schmidt, A. Beutler and B. Snovydovych, Eur. J. Inorg. Chem., 2008, 4073–4095 CrossRef.
- M.-H. Lin, H.-J. Liu, W.-C. Lin, C.-K. Kuo and T.-H. Chuang, Org. Biomol. Chem., 2015, 13, 11376–11381 RSC.
- N. Cankařová, J. Hlaváč and V. Krchňák, Org. Prep. Proced. Int., 2010, 42, 433–465 CrossRef.
- H. Yoshida, Y. Mimura, J. Ohshita and A. Kunai, Chem. Commun., 2007, 2405–2407 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: NMR (1H and 13C), IR, LRMS (ESI) and HRMS (ESI) of all featured compounds. See DOI: 10.1039/c8cc06556j |
| This journal is © The Royal Society of Chemistry 2018 |