
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient cosubstrate enzyme pairs for sequence-specific methyltransferase-directed photolabile caging of DNA†
Michael
Heimes
 ,
Leonie
Kolmar
and
Clara
Brieke
*
,
Leonie
Kolmar
and
Clara
Brieke
*
Department of Biomolecular Mechanisms, Max Planck Institute for Medical Research, Jahnstrasse 29, D-69120 Heidelberg, Germany. E-mail: Clara.Brieke@mpimf-heidelberg.mpg.de
First published on 19th October 2018
Abstract
Supplemented with synthetic surrogates of their natural cosubstrate S-adenosyl-L-methione (AdoMet), methyltransferases represent a powerful toolbox for the functionalization of biomolecules. By employing novel cosubstrate derivatives in combination with protein engineering, we show that this chemo-enzymatic method can be used to introduce photolabile protecting groups into DNA even in the presence of AdoMet. This approach enables optochemical control of gene expression in a straight-forward manner and we have termed it reversible methyltransferase directed transfer of photoactivatable groups (re-mTAG).
The use of photolabile protecting groups (“caging groups”) in biochemical research to trigger the activity of a bioactive molecule with light has enabled complex investigations with unprecedented spatiotemporal resolution. It has been very powerful for controlling activity of small molecules including metabolites or lipids in vitro and in vivo and has been expanded to larger biomolecules such as proteins and nucleic acids.1,2 Still, the site-specific introduction of caging groups into complex biomolecules remains challenging. While the use of unnatural amino acids by the extension of the genetic code nowadays enables site-specific caging of proteins without size limitation in living cells,3,4 such a methodology has not been developed for nucleic acids so far. Herein, we present a chemo-enzymatic strategy that has the potential to overcome these limitations.
Photoprotected nucleic acids can be prepared either postsynthetically or by introduction of a modified nucleoside into DNA or RNA by means of solid-phase synthesis. While postsynthetic methods for caging nucleic acids typically make use of highly reactive diazo compounds leading to randomized caging patterns with the difficulty of incomplete (un)caging,5–8 the latter approach allows a site-specific modification and thus enables effective regulation of various processes such as transcription or gene expression.9–13 Still, it relies on extensive synthetic organic chemistry and its adaption for the photocontrol of plasmids or large RNA constructs can be cumbersome.
Taking advantage of the inherent target specificity of enzymes, chemo-enzymatic methods depict another strategy to introduce modifications into complex biomolecules and have already proven valuable for labelling various classes of biomolecules in vitro and in vivo. Especially S-adenosyl-L-methionine (AdoMet)-dependent methyltransferases (MTases) represent a valuable toolbox for targeted nucleic acid labelling by using promiscuous or engineered MTases that are able to catalyse the transfer of functional moieties onto their target molecule employing so-called “doubly-activated” derivatives of the native cosubstrate AdoMet.14–16 This MTase-directed transfer of activated groups (mTAG)17 has been used for single-molecule optical DNA mapping,18 tagging of epigenomic target sites19 as well as RNA labelling20,21 and has even been used to probe histone and RNA Mtases in living cells.22,23 For this purpose, various AdoMet analogues with extended side-chains bearing transferable alkene, alkyne, amino, 4-vinyl benzyl and very recently photoactivatable moieties have been developed.16,24,25
To overcome the issues inherent to current methods for caging DNA, we herein have addressed the potential of mTAG for the photoregulation of DNA. Thus, we decided to explore the enzymatic transfer of photoactivatable nitrobenzyl (NB) groups using the adenine-N6 MTase M.TaqI from Thermus aquaticus. M.TaqI is well-studied and known to be promiscuous,14,18 while NB groups are widely applied caging groups.1 We designed four AdoMet analogues bearing transferable NB groups differing in their methoxy substitution pattern of the NB core (see Fig. 1). While such substitutions are known to increase the kinetics of photocleavage,26 we further show that they permit us to modulate the efficiency of the enzymatic transfer reaction. Complemented by mutagenesis of the cosubstrate binding site of M.TaqI we were able to generate cosubstrate enzyme pairs which catalyse DNA caging with high efficiency despite presence of the native cosubstrate AdoMet. Finally, we were intrigued to explore the utility of enzymatic DNA caging to control gene expression.
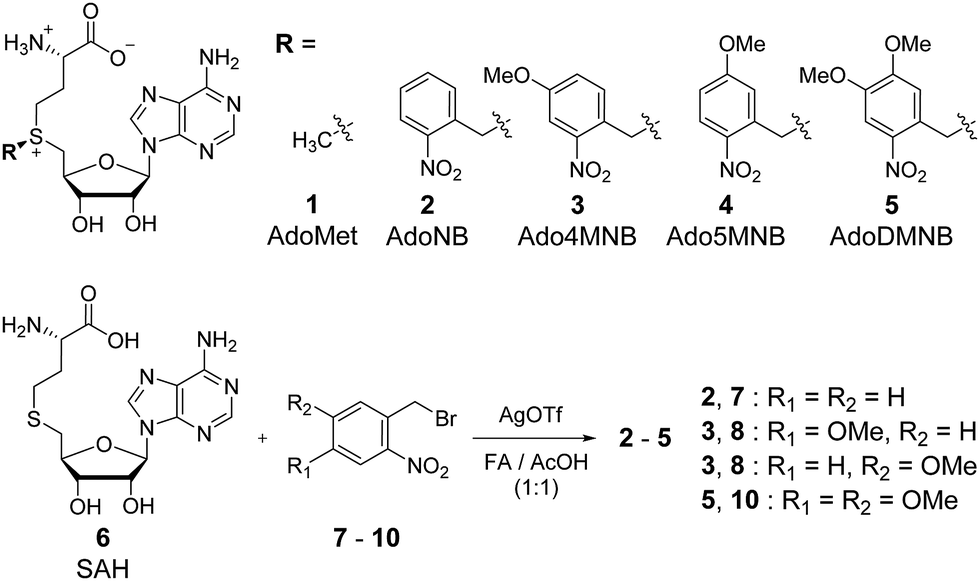 | ||
| Fig. 1 Structures of cosubstrates used in this study and synthesis conditions. | ||
The NB derivatives 2–5 were synthesized by alkylation of S-adenosylhomocysteine (SAH, 6) with the respective halides (7–10) using reported conditions.14 Purification and isolation of the enzymatically active (S)-diastereomers using preparative reversed-phase HPLC was confirmed by HPLC, MS and 1H-NMR. Half-lives for 2–5 were determined in the range of 3–4 hours under physiological conditions (Fig. S5, ESI†) suitable for enzymatic reactions and comparable with previously reported data.24,27
To evaluate MTase-directed caging as well as the uncaging reaction in more detail, we developed an in vitro activity assay. Shortly, a hemi-methylated duplex ODNI·ODNIIMe (Fig. 2a) containing the palindromic M.TaqI recognition sequence 5′-TCGA-3′ was incubated with M.TaqI and cosubstrate under saturating conditions (Fig. S15, ESI†) The reaction was monitored using ion-pairing reversed-phase UHPLC-MS analysis at elevated temperatures to separate both strands which allowed identification as well as quantification of the reaction products. These assays revealed that M.TaqI accepts not only AdoNB (2) – as it was recently reported by Rentmeister et al.25 – but also catalyses the transfer of extended NB groups fast and efficiently within minutes. Alkylation with Ado4MNB (3) and AdoDMNB (5) even proceeds at rates comparable to the native substrate AdoMet (1) (Fig. 2b and c). This is remarkable considering that the transalkylation rates of M.TaqI we and others have observed for allylic cosubstrates are usually at least one order of magnitude smaller than the equivalent methyl transfer14 (Fig. S12, ESI†). A cause for this effect may be the enhanced reactivity of benzylic positions towards substitution and the conjugative stabilization of the sp2-hybridized SN2-transition state through the aromatic system, which has recently also been observed for RNA MTases.28
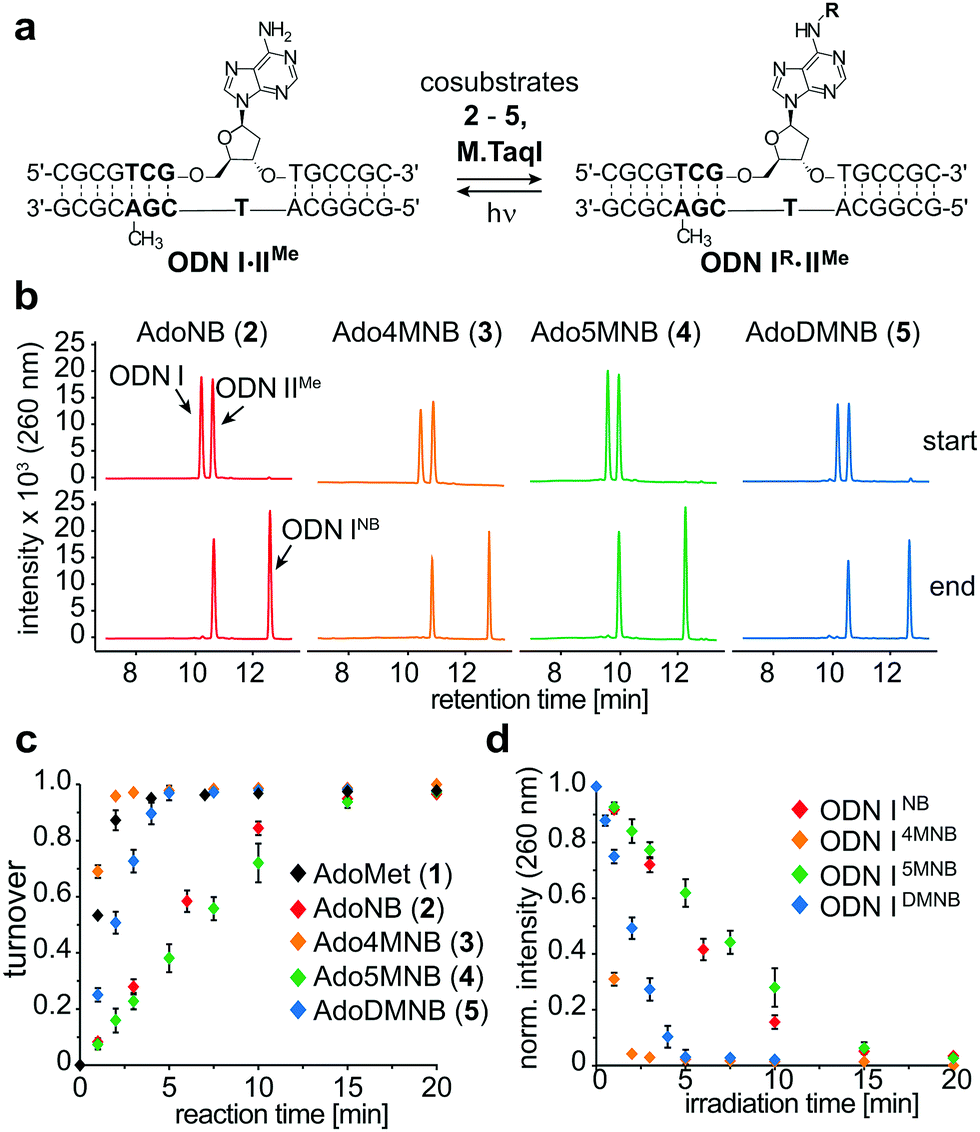 | ||
| Fig. 2 In vitro activity assay for characterizing M.TaqI-catalyzed NB-transfer onto DNA. (a) Duplex used for this assay (R = NB, 4MNB, 5MNB, DMNB); (b) representative UHPLC chromatograms of enzymatic transfer reaction with cosubstrates 2–5 at t = 0 min and 15 min; (c) time course experiments for enzymatic transfer of 1–5 (500 μM) onto ODNI·ODNIIMe (25 μM) by M.TaqI (2 μM) at 37 °C; (d) time series of uncaging NB-labeled ODNIR using light at 365 nm; (e) competition assays with increasing amounts of cosubstrates 2–5 (500 μM–4 mM) in the presence of 500 μM AdoMet (1). Experiments were performed in triplicates. | ||
Irradiating samples of caged duplex ODNIR·ODNIIMe for different time periods at λ = 365 nm using a UV hand lamp (6 W) revealed clean and rapid photorelease of uncaged duplex for NB-, 4MNB- and 5MNB-modified ODNI and thus confirmed the reversibility of the enzymatic labelling reaction (Fig. 2d and Fig. S13, ESI†). Merely for ODNIDMNB·ODNIIMe, we were able to observe the formation of side products due to reaction with the respective nitrosoaldehyde.
Encouraged by these results, we addressed the question if cosubstrates 2–5 are able to compete with AdoMet for M.TaqI, thus potentially permitting enzymatic caging of DNA in a cellular context. Hence, we performed alkylation assays under steady state conditions with varying ratios of cosubstrate analogue to AdoMet (Fig. 3a). These experiments revealed striking differences between the NB derivatives; while AdoMet (1) was clearly favoured over the unsubstituted AdoNB (2), the differences in enzymatic transfer efficiency observed between 1 and 3–5 became less with AdoDMNB (5) being nearly equally efficiently utilized by M.TaqI as AdoMet (1). Thus, the excess of cosubstrate surrogate needed to outcompete AdoMet reduced from approximately five-fold for AdoNB (2) to 1.5-fold for AdoDMNB (5) indicating that enzymatic caging of DNA can proceed despite the presence of AdoMet.
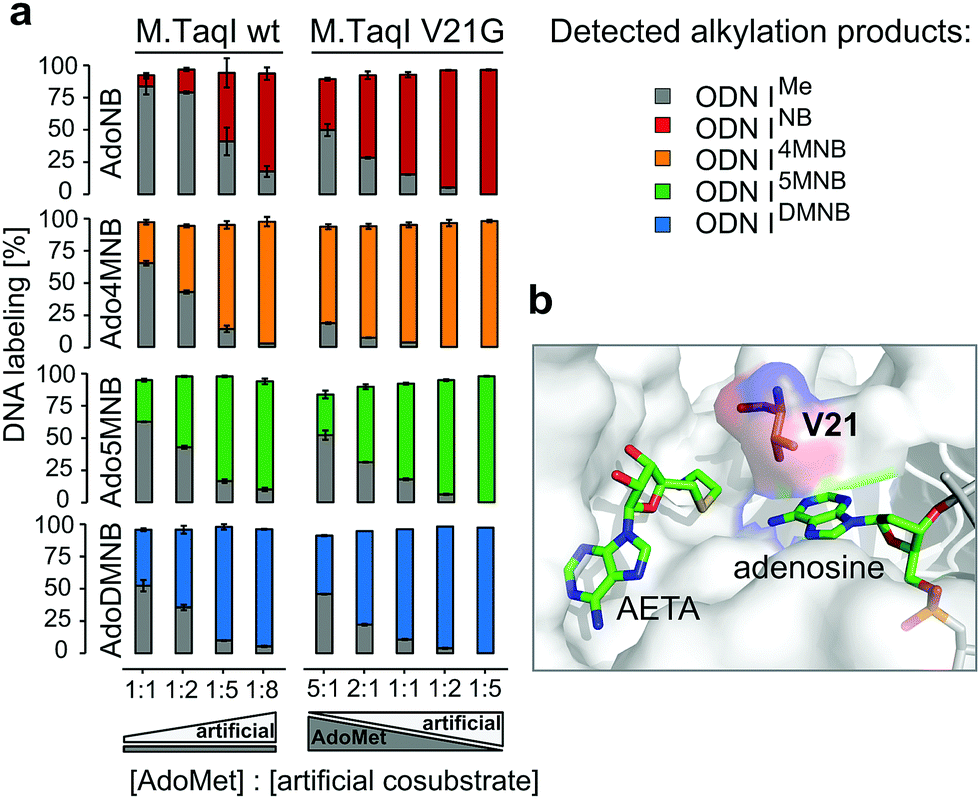 | ||
| Fig. 3 (a) Cosubstrate competition experiments with either wild-type M.TaqI or M.TaqI V21G: in the experiments with wild-type M.TaqI the concentration of cosubstrates 2–5 (500 μM–4 mM) was varied in the presence of 500 μM AdoMet (1), while with M.TaqI V21G the amounts of cosubstrates 2–5 and AdoMet (1) were varied (500 μM, 1 mM, 2.5 mM); (b) cosubstrate binding site of M.TaqI with highlighted V21 (PDB ID 1g38; AETA: nonreactive AdoMet analog (5′-[2-(amino)ethylthio]-5′-deoxyadenosine)). Experiments were done in triplicates. | ||
Steric engineering of the cosubstrate binding pocket represents another approach to further increase the efficiency of MTase-catalyzed reactions with artificial cosubstrates.29 Albeit wild-type M.TaqI is already known to be very promiscuous and employs NB-derived cosubstrates with high efficiency, we became intrigued to revisit the crystal structure (PDB: 1g38) of M.TaqI. Hereby, we concluded that V21, which is in close proximity to the sulfur atom of the cosubstrate, might sterically interfere with extended NB groups (Fig. 3b).
Thus, we generated a single-residue mutant of M.TaqI by replacing V21 with glycine and subsequently analysed the reactivity profile of M.TaqI V21G with our established in vitro activity assay using artificial cosubstrates 2–5 as well as AdoMet. Interestingly, transfer rates for the artificial cosubstrates remained mainly unaffected by this mutation indicating that NB groups can be accommodated in the binding pocket without steric clashes. Only for Ado5MNB (4) the transalkylation rate was slightly increased (Fig. S16, ESI†). In contrast to that, we observed a pronounced effect on the rate for methyl transfer, which was slowed down over six-fold and we assume that V21 may play a role in correctly positioning AdoMet in the cosubstrate binding pocket or contributes to its binding affinity via hydrophobic interactions. This significant drop in transmethylation rate prompted us to repeat competition assays with varying ratios of cosubstrate analogue to AdoMet using M.TaqI V21G. And indeed, this mutant demonstrated a strong preference for NB-derived cosubstrates even with AdoMet being present in excess (Fig. 3a). Thus, even at two-fold excess of AdoMet, transalkylation was catalysed at least twice as efficient than transmethylation by M.TaqI V21G and at equimolar amounts of artificial and native cosubstrate the excess of detected NB-modified ODNI over methylated ODNI ranged from around four-fold (AdoNB (2) and Ado5MNB (4)) up to 25-fold for AdoDMNB (5) and Ado4MNB (3). Especially Ado4MNB (3) was employed with superior efficiency by M.TaqI V21G outcompeting AdoMet even if this was present in five-fold excess. Thus, with M.TaqI V21G and our NB-derived cosubstrates we have identified MTase cosubstrate pairs which enable site-specific enzymatic caging of DNA in the presence of AdoMet. This is an essential prerequisite for applications in a biological context and strengthens the potential of re-mTAG as an approach for caging DNA in living cells.
To probe the applicability of re-mTAG in a biochemical context, we studied the photoregulation of gene expression exemplified by sfYFP (sfYFP: superfolder yellow fluorescent protein). For that, we made use of three different expression plasmids: construct V1 represents a pET28 vector encoding for sfYFP under the control of the T7 promoter. Since the sfYFP gene itself contains 6 M.TaqI recognition sites distributed over the whole gene (Fig. 4a and Fig. S18, ESI†), we were interested if caging these sites would already enable photoregulation of sfYFP expression. However, since the T7 RNA polymerase (T7 RNAP) is known to form a very stable elongation complex with the template strand during transcription capable of bypassing lesion sites,30 we further designed plasmids featuring two (V2) or one (V3) additional M.TaqI recognition sites in close proximity downstream to the T7 promoter region (Fig. 4a and Fig. S19, S22, ESI†).
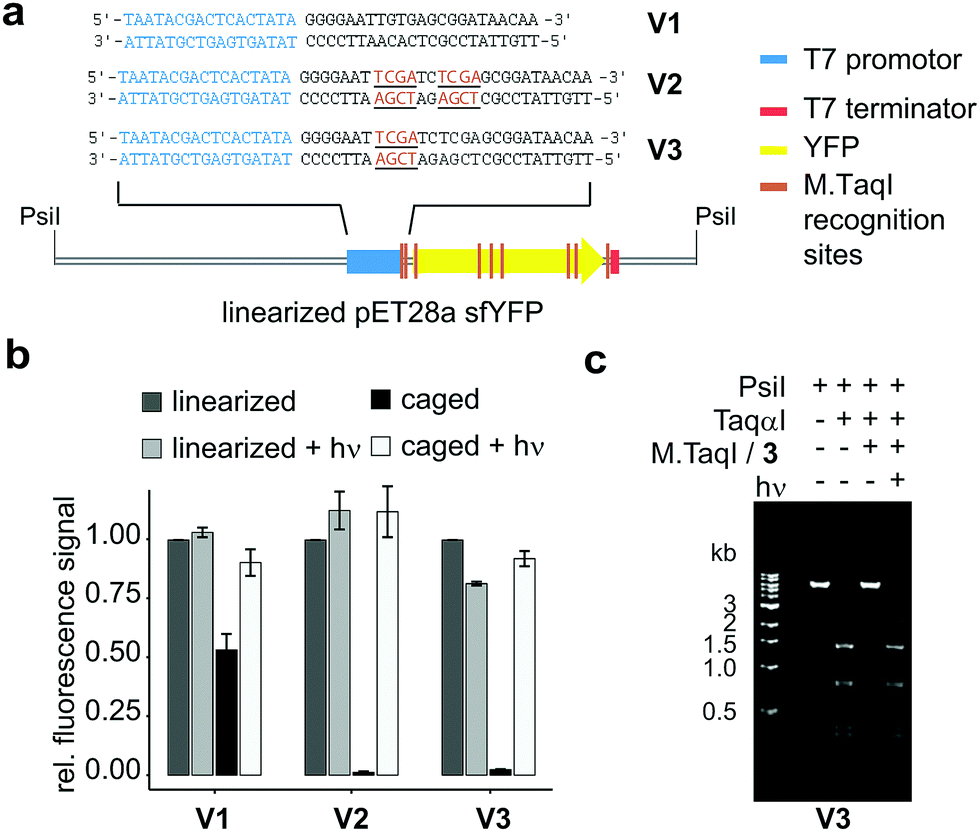 | ||
| Fig. 4 In vitro transcription/translation assay; (a) constructs used with M.TaqI recognition sites highlighted; (b) relative amounts of sfYFP expressed after 2 h with or without initial irradiation (LED, 5 min, 365 nm), experiments were performed in triplicates; (c) modification restriction assay confirming successful caging and uncaging of linearized plasmid V3 (details see ESI†). | ||
The plasmids were caged using M.TaqI and Ado4MNB and completeness as well as reversibility of the labelling reaction was confirmed by performing restriction assays with the corresponding restriction endonuclease TaqαI (Fig. 4c and Fig. S17, ESI†). Caged and control plasmids were linearized using PsiI and included as templates in in vitro expression assays (for details see ESI†). In-gel fluorescence of the translated sfYFP protein was utilized as a quantitative readout to evaluate the caging groups’ effect on gene expression.
Our results demonstrate that re-mTAG does indeed enable photoregulation of gene expression and furthermore highlight the importance of site-specific caging (Fig. 4b). In previous studies, the promoter region itself has been identified as suitable caging site for efficient photoregulation of gene expression by preventing the RNAP from recognizing the promoter region.5,7 Here, we show that introducing caging groups in close proximity downstream to the T7 promotor also results in full – and upon illumination reversible – silencing of transcription, while random caging of the sfYFP gene only partially suppresses gene expression indicating that the T7 RNAP can efficiently transcribe past 4MNB-modified sites (Fig. S21, ESI†). The fact, that already a single 4MNB group located in proximity to the promoter either on coding or noncoding strand (see Fig. S7 and S8, ESI†) aborts transcription completely, is remarkable and we assume that the caging group interferes with the transition of the T7 RNAP from initiation to the highly processive elongation phase. This transition takes place after the nascent RNA is extended to 8–12 nucleotides and represents a critical multistep process accompanied by conformational changes in both RNAP and DNA.31 Albeit M.TaqI targets further sides in the expression vector, gene expression can be restored to its initial value upon illumination demonstrating clean uncaging of the enzymatically caged plasmid and thus resolves a major issue inherent to stochastically caged plasmids.
In conclusion, reversible methyltransferase directed transfer of photoactivatable groups (re-mTAG) represents a powerful enzymatic method to introduce caging groups site-specifically into large DNA molecules and we have demonstrated its feasibility for controlling plasmid-based gene expression using light. We have shown that the methyltransferase M.TaqI acts as a highly potent biocatalyst for photolabelling not only by accepting a variety of nitrobenzyl-modified cosubstrate analogues but also by transferring such groups at comparable rates as the native AdoMet. Furthermore, utilizing protein engineering we have generated an M.TaqI mutant that features a strong preference for NB-derived cosubstrates over AdoMet enabling caging of DNA in the presence of native cofactor with high selectivity. The developed cosubstrate enzyme pairs have the potential to extent the applicability of re-mTAG to environments even as competitive as those inside living cells. In the light of recent discoveries regarding the in vivo synthesis of AdoMet derivatives using cell-permeable methionine analogues22,23,32 and regarding the continuous need for methods that enable manipulation of processes in living cells, we anticipate a broad range of applications for this method in the near future.
This work was supported by the Max Planck Society. The authors thank M. Müller and S. Fabritz for technical support and J. Reinstein, A. Meinhart and E. Weinhold for discussions. C. B. thanks I. Schlichting (MPI-Hd) for her constant support.
Open Access funding provided by the Max Planck Society.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2012, 51, 8446–8476 CrossRef CAS PubMed.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed.
- S. M. Hancock, R. Uprety, A. Deiters and J. W. Chin, J. Am. Chem. Soc., 2010, 132, 14819–14824 CrossRef CAS PubMed.
- M. J. Schmidt and D. Summerer, Front. Chem., 2014, 2, 2296–2646 Search PubMed.
- W. T. Monroe, M. M. McQuain, M. S. Chang, J. S. Alexander and F. R. Haselton, J. Biol. Chem., 1999, 274, 20895–20900 CrossRef CAS PubMed.
- H. Ando, T. Furuta, R. Y. Tsien and H. Okamoto, Nat. Genet., 2001, 28, 317–325 CrossRef CAS PubMed.
- S. Shah, S. Rangarajan and S. H. Friedman, Angew. Chem., Int. Ed., 2005, 44, 1328–1332 CrossRef CAS PubMed.
- T. Stafforst and J. M. Stadler, Angew. Chem., Int. Ed., 2013, 52, 12448–12451 CrossRef CAS PubMed.
- L. Kröck and A. Heckel, Angew. Chem., Int. Ed., 2005, 44, 471–473 CrossRef PubMed.
- X. Tang and I. J. Dmochowski, Mol. BioSyst., 2007, 3, 100–110 RSC.
- J. Hemphill, J. Govan, R. Uprety, M. Tsang and A. Deiters, J. Am. Chem. Soc., 2014, 136, 7152–7158 CrossRef CAS PubMed.
- S. Yamaguchi, Y. Chen, S. Nakajima, T. Furuta and T. Nagamune, Chem. Commun., 2010, 46, 2244–2246 RSC.
- X. Wang, M. Feng, L. Xiao, A. Tong and Y. Xiang, ACS Chem. Biol., 2016, 11, 444–451 CrossRef CAS PubMed.
- C. Dalhoff, G. Lukinavičius, S. Klimasauskas and E. Weinhold, Nat. Chem. Biol., 2006, 2, 31–32 CrossRef CAS PubMed.
- S. Klimašauskas and E. Weinhold, Trends Biotechnol., 2007, 25, 99–104 CrossRef PubMed.
- J. Deen, C. Vranken, V. Leen, R. K. Neely, K. P. F. Janssen and J. Hofkens, Angew. Chem., Int. Ed., 2017, 56, 5182–5200 CrossRef CAS PubMed.
- G. Lukinavičius, V. Lapienė, Z. Staševskij, C. Dalhoff, E. Weinhold and S. Klimašauskas, J. Am. Chem. Soc., 2007, 129, 2758–2759 CrossRef PubMed.
- S. Kim, A. Gottfried, R. R. Lin, T. Dertinger, A. S. Kim, S. Chung, R. A. Colyer, E. Weinhold, S. Weiss and Y. Ebenstein, Angew. Chem., Int. Ed., 2012, 51, 3578–3581 CrossRef CAS PubMed.
- E. Kriukienė, V. Labrie, T. Khare, G. Urbanavičiūtė, A. Lapinaitė, K. Koncevičius, D. Li, T. Wang, S. Pai, C. Ptak, J. Gordevičius, S.-C. Wang, A. Petronis and S. Klimašauskas, Nat. Commun., 2013, 4, 2190 CrossRef PubMed.
- A. Plotnikova, A. Osipenko, V. Masevičius, G. Vilkaitis and S. Klimašauskas, J. Am. Chem. Soc., 2014, 136, 13550–13553 CrossRef CAS PubMed.
- J. M. Holstein, L. Anhäuser and A. Rentmeister, Angew. Chem., Int. Ed., 2016, 55, 10899–10903 CrossRef CAS PubMed.
- R. Wang, K. Islam, Y. Liu, W. Zheng, H. Tang, N. Lailler, G. Blum, H. Deng and M. Luo, J. Am. Chem. Soc., 2013, 135, 1048–1056 CrossRef CAS PubMed.
- K. Hartstock, B. Nilges, A. Ovcharenko, N. Cornelissen, N. Puellen, S. Leidel and A. Rentmeister, Angew. Chem., Int. Ed., 2018, 57, 6342–6346 CrossRef CAS PubMed.
- F. Muttach, F. Mäsing, A. Studer and A. Rentmeister, Chem. – Eur. J., 2017, 23, 5988–5993 CrossRef CAS PubMed.
- L. Anhäuser, F. Muttach and A. Rentmeister, Chem. Commun., 2018, 54, 449–451 RSC.
- C. P. Holmes, J. Org. Chem., 1997, 62, 2370–2380 CrossRef CAS PubMed.
- G. Lukinavičius, M. Tomkuvienė, V. Masevičius and S. Klimašauskas, ACS Chem. Biol., 2013, 8, 1134–1139 CrossRef PubMed.
- F. Muttach, N. Muthmann, D. Reichert, L. Anhäuser and A. Rentmeister, Chem. Sci., 2017, 8, 7947–7953 RSC.
- G. Lukinavičius, A. Lapinaitė, G. Urbanavičiūtė, R. Gerasimaitė and S. Klimašauskas, Nucleic Acids Res., 2012, 40, 11594–11602 CrossRef PubMed.
- C. You, P. Wang, X. Dai and Y. Wang, Nucleic Acids Res., 2014, 42, 13706–13713 CrossRef CAS PubMed.
- R. P. Bandwar, N. Ma, S. A. Emanuel, M. Anikin, D. G. Vassylyev, S. S. Patel and W. T. McAllister, J. Biol. Chem., 2007, 282, 22879–22886 CrossRef CAS PubMed.
- S. Singh, J. Zhang, T. D. Huber, M. Sunkara, K. Hurley, R. D. Goff, G. Wang, W. Zhang, C. Liu, J. Rohr, S. G. Van Lanen, A. J. Morris and J. S. Thorson, Angew. Chem., 2014, 126, 4046–4050 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Biochemical and synthetic procedures, HPLC and analytical data. See DOI: 10.1039/c8cc05913f |
| This journal is © The Royal Society of Chemistry 2018 |