
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
1,1-Diphosphines and divinylphosphines via base catalyzed hydrophosphination†
N. T.
Coles
,
M. F.
Mahon
and
R. L.
Webster
 *
*
Department of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: r.l.webster@bath.ac.uk
First published on 22nd August 2018
Abstract
A catalytic hydrophosphination route to 1,1-diphosphines is yet to be reported: these narrow bite angle pro-ligands have been used to great effect as ligands in homogeneous catalysis. We herein demonstrate that terminal alkynes readily undergo double hydrophosphination with HPPh2 and catalytic potassium hexamethyldisilazane (KHMDS) to generate 1,1-diphosphines. A change to H2PPh leads to the formation of P,P-divinyl phosphines.
Bidentate phosphines constitute an immensely important class of compounds used throughout synthetic chemistry, where they are commonly employed as ligands in transition metal catalysis.1–3 An array of diphosphines have been prepared and the effect of the phosphine bite angle has been studied in catalysis.4 Narrow bite-angle 1,1-diphosphines, whereby the phosphorus is linked by a single methylene (or methine) group, have been used with great success in catalytic hydrofunctionalization,5–13 hydrogenation14–16 and epoxidation,17–19 to name but three reaction classes. However, unlike many ligand series used in catalysis, reports of a truly diverse homologous series of 1,1-diphosphine compounds are scarce.20 Commercially available examples of the free ligand are limited to bis(dimethylphosphino)methane (DMPM), bis(dicyclohexylphosphino)methane and bis(diphenylphosphino)methane (DPM). The range of functionality that can be tolerated is linked to the challenges associated with the synthesis of such pro-ligands: commonly reaction of the deprotonated phosphine with a dihalomethane is necessary (Scheme 1a).21–24 The α-deprotonation of a methyl-diaryl/alkyl phosphine and subsequent reaction with a chlorophosphine, is also known (Scheme 1b).25–27 Both methods are synthetically wasteful, but the latter can give access to unsymmetrically substituted 1,1-diphosphines.
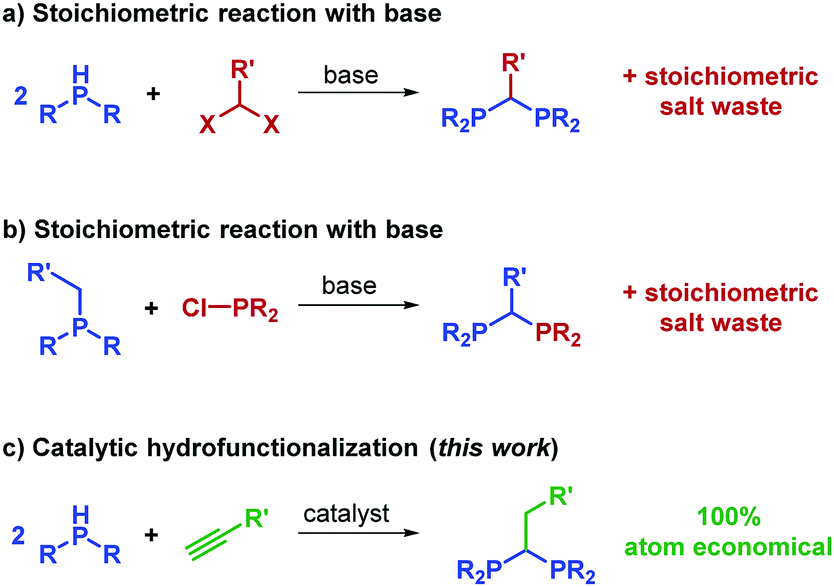 | ||
| Scheme 1 1,1-Diphosphines can be formed from stoichiometric reaction of an organohalide, phosphine and base whereas this work shows that they can also form using catalytic hydrophosphination chemistry. | ||
The stoichiometric reaction of a base with phosphines and alkynes has been known for many years.28,29 One of the first examples where a base was used catalytically (25 mol% loading of PhLi) was demonstrated by Märkl and Potthast to prepare phospholes.30 Bookham and Smithies subsequently performed a comprehensive study regarding hydrophosphination of non-terminal alkynes using 22 mol% KOtBu as the catalyst, but with the addition of NEt3 as a solvent.31 The limitations of this reaction lie not only in the fact that only three or four catalytic turnovers per potassium centre were achieved, but also in the lack of selectivity obtained: the phosphinoalkene product was obtained as a mixture of E and Z isomers and often the reaction was contaminated with tetraphenyldiphosphine, (Ph2P)2.
We herein report a mild, facile and selective route to a homologous series of 1,1-diphosphines using alkynes and a catalytic amount of KHMDS (Scheme 1c). The most similar reported methodology and range of products of which we are aware has come from Leung. Using ethyl propiolate and but-3-yn-2-one, a chiral template approach was employed by Leung giving a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mixture of the stereoisomeric 1,1-diphosphine ligated Pd complexes.32 Interestingly, the 1,1-diphosphine Pd-complex was formed using catalytic amounts of NEt3 but the 1,2-diphosphine Pd-complex was formed in the presence of excess NEt3. HCl (conc.) followed by KCN (aq.) washes were necessary to release the phosphine from the Pd template.
:2 mixture of the stereoisomeric 1,1-diphosphine ligated Pd complexes.32 Interestingly, the 1,1-diphosphine Pd-complex was formed using catalytic amounts of NEt3 but the 1,2-diphosphine Pd-complex was formed in the presence of excess NEt3. HCl (conc.) followed by KCN (aq.) washes were necessary to release the phosphine from the Pd template.
Following a short optimization procedure using ethyl propiolate as our standard substrate, we found that activated alkynes can readily undergo base catalyzed double hydrofunctionalization to yield 1,1-diphosphines at room temperature without the need for any reagents in excess. Weak bases such as K2CO3 or NEt3 do not achieve good selectivity or yield. As an inexpensive and easy to handle base, 10 mol% NaOtBu gives quantitative spectroscopic yield of 1b in 6 h. Comparable spectroscopic yields of 1b are also achieved in 6 h using KOtBu, NaHMDS and KHMDS. When the loading of these bases is lowered to 1 mol% it is still possible to achieve very high conversion (>90%, see ESI† for base optimisation). After 16 h at RT in the absence of any base 7% 1b is obtained along with 49% vinyl phosphine (3:1 mixture of E and Z isomers) and unreacted HPPh2. When we proceeded to investigate reactivity with other alkynes, we found that NaOtBu is not a suitable catalyst. For example, 3-ethynylpyridine only generates 1o in 17% spectroscopic yield with NaOtBu, a change of alkali metal to KOtBu increases the yield to 38%, but it is only when a change to KHMDS is exercised that a high spectroscopic yield of 1o is obtained (92%). This is presumably linked to the non-nucleophilic character of KHMDS which leads to cleaner reactivity (multiple side-products are formed from the reaction of NaOtBu and KOtBu). This trend is observed across other classes of substrate: see ESI† for reaction monitoring using NaOtBu, KOtBu and KHMDS with each class of substrate. Hydrophosphination does not proceed in CH2Cl2, C6H6 or C6H5F and thus we investigated substrate scope using 10 mol% KHMDS in MeCN (Scheme 2).
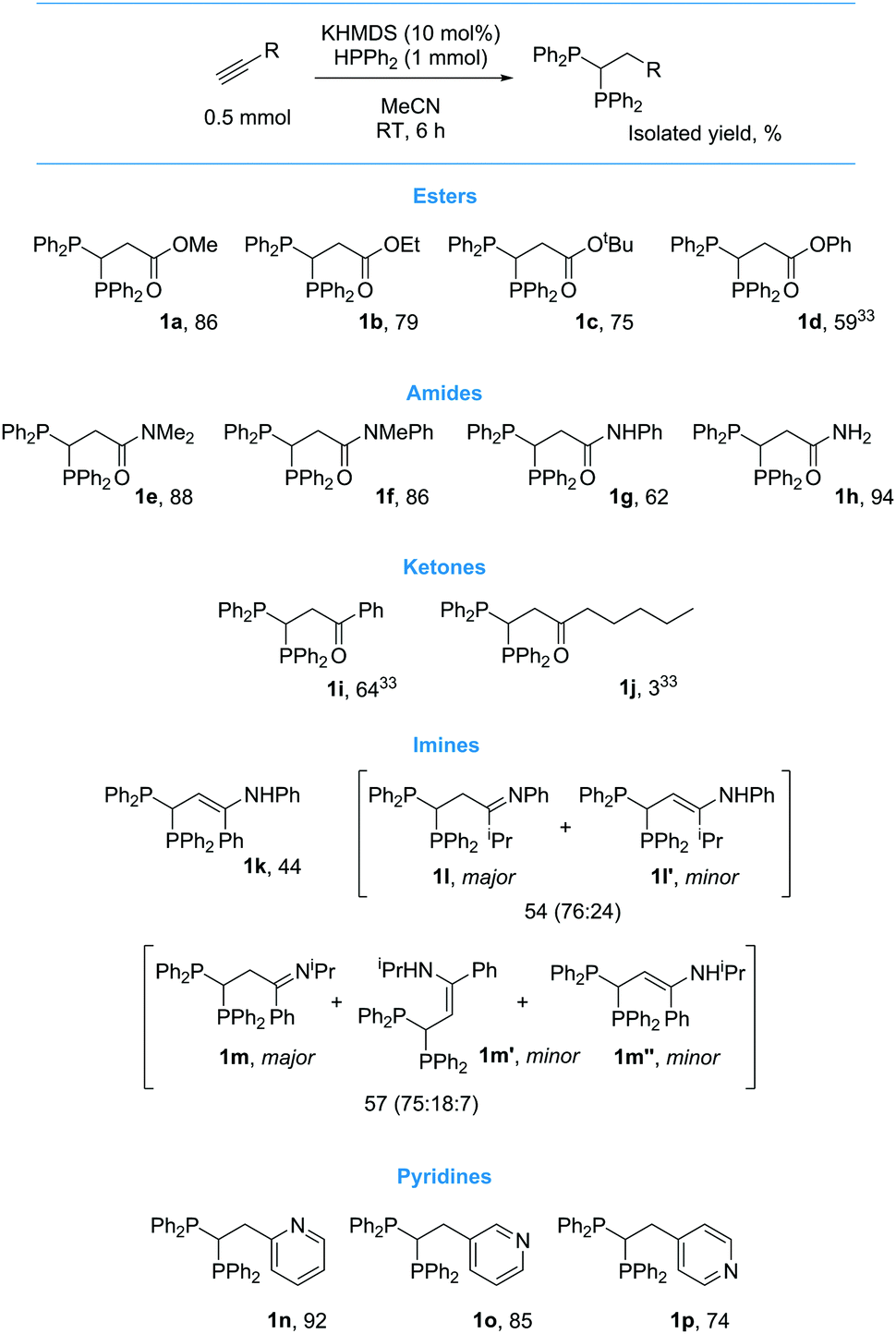 | ||
| Scheme 2 1,1-Diphosphines formed from the reaction of functionalized acetylenes with HPPh2 in the presence of 10 mol% KHMDS. | ||
In general, the reaction appears to benefit from carbonyl functionality α to the alkyne and thus other propiolates give high yield of the desired product (1a to 1d33). Both tertiary (1e and 1f), secondary (1g) and primary (1h) propargyl amides and aromatic ketones (1i33) have been functionalized with good isolated yield. Aliphatic ketones give low yield of product (1j33). Imines also react well but undergo concomitant tautomerization to give the enamine (1k to 1m/1m′/1m′′). We are pleased to report that ethynylpyridines can be used in the transformation and that the substitution pattern has little bearing on the level of reactivity observed (1n to 1p). Unfortunately, the reaction is not as clean when other aryl acetylenes are employed.34
Crystals of 1b, 1f, 1i and 1k were isolated (see Fig. 1 for 1b and ESI† for 1f, 1i and 1k). The compounds all have fairly disparate P–C–P bond angles. This is, presumably, linked to sterics of the pendant functional group with the tertiary amide of 1f providing the greatest steric bulk and therefore forcing the P–C–P bond angle to become more acute (99.41(9)°) than that observed for the ester (1b: 103.60(8)°), whereas the phenyl ketone (1i) has a wider P–C–P bond angle at 105.75(9)°. These P–C–P bond angles are in-line with that of DPM,35 which is reported as 106.17°. Enamine 1k has a wider P–C–P bond angle of 108.53(8)°. It can be surmised that these crystal data indicate that our hydrofunctionalization products are primed for use as ligands in transition metal complexes.
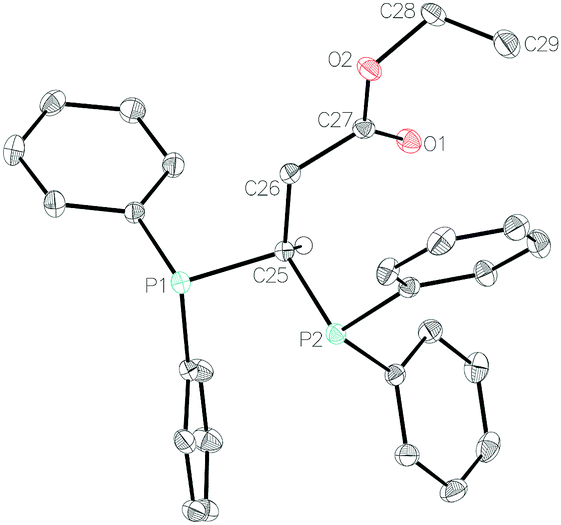 | ||
| Fig. 1 Molecular structure of 1b. Ellipsoids are represented at 30%. With the exception of the methine proton, all hydrogen atoms have been omitted for clarity. | ||
A radical clock study of our standard reaction (0.5 mmol ethyl propiolate, 1 mmol HPPh2, 10 mol% KHMDS, 1 mL MeCN with 1 mmol (chloromethyl)cyclopropane) gives 70% 1b after 1 h and 98% 1b after 6 h i.e. results that are comparable to those obtained in the absence of a radical clock34 suggesting a non-radical mediated reaction. During our reaction monitoring studies a small quantity of the E-vinyl phosphine moiety is observed by in situ NMR spectroscopy after 1 h suggesting that this is the (sterically driven) product of the first hydrophosphination and the Michael accepting properties of this phosphinoalkene then facilitate the regioselective second hydrophosphination reaction. To further investigate the role of the base, ethyl propiolate-d was used in a catalytic reaction. Deuterium is observed at both the methine and methylene positions of 1b-d. Order of addition of reagents/catalyst do not appear to impact on the reaction. HPPh2 and ethyl propiolate-d do not undergo H/D exchange in the absence of KHMDS. Given the acidity of the propargylic proton, we believe that competitive deprotonation of this moiety takes place which can lead to deuteration of both methylene and methine carbons (Scheme 3).
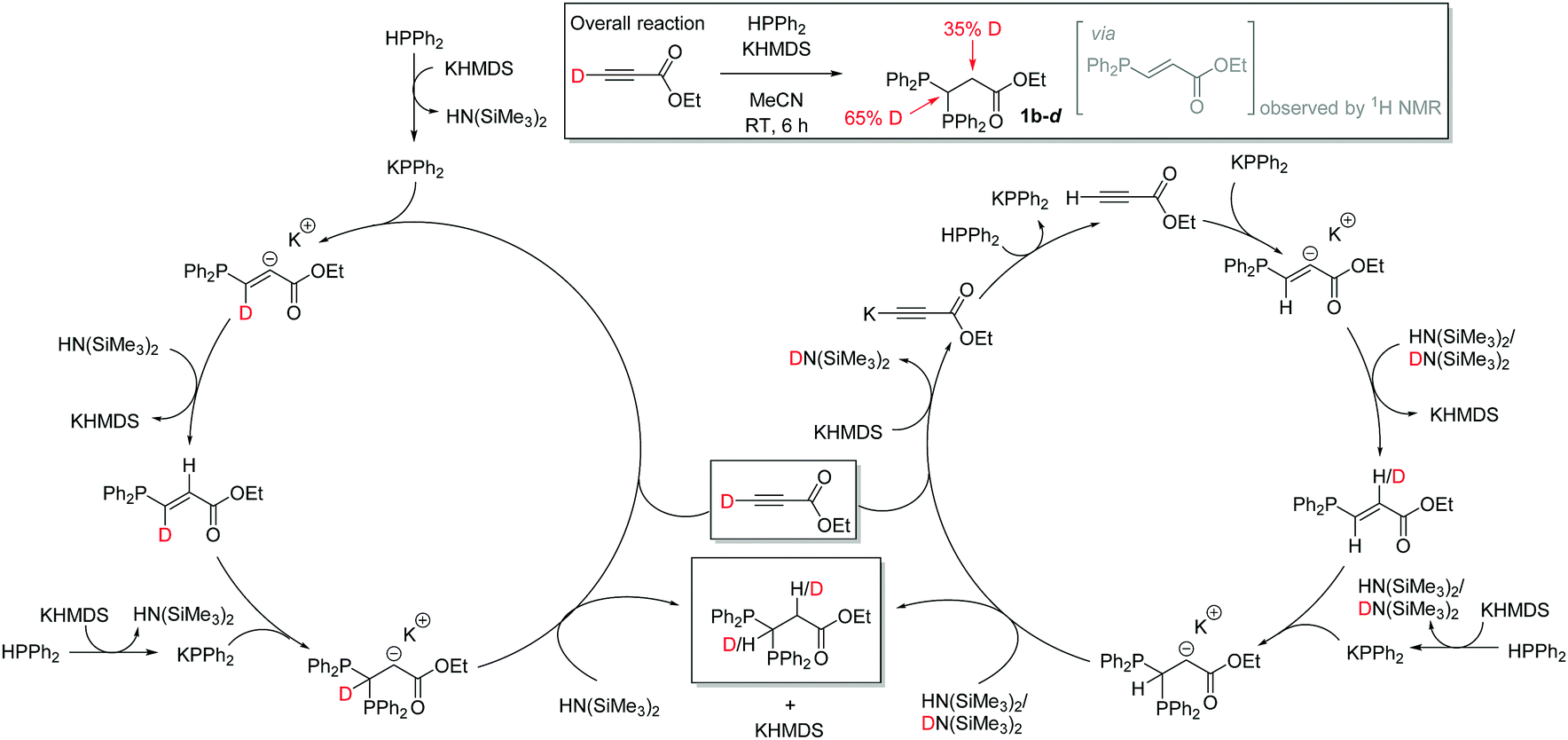 | ||
| Scheme 3 Results of deuterium labelling study and proposed catalytic cycle, with potential route by which deuteration could occur at the methylene position (right hand cycle). | ||
By changing the phosphine to a primary phosphine (H2PPh), we are able to afford divinylphosphines in the Z,Z configuration, with only one exception (Scheme 4). P,P-Divinyl phosphines have only previously been reported by Issleib using the potassium salt of phenylphosphine and appeared to give a mixture of isomers,29 or by Kobayashi who used AIBN but obtained a mixture of isomers (52% Z,Z, 30% Z,E and 18% E,E).36 More recently Takahashi used cross-coupling, rather than a hydrofunctionalization approach, employing phenyl- and n-hexyl-vinylzirconocene reagents (where the E-geometry was predefined) and dichlorophenylphosphine to afford the E,E divinylphosphine product.37 The transformation we report here complements the Zr-catalyzed method of Waterman, whereby internal alkynes undergo mono-hydrophospination with H2PPh to yield a mixture of E- and Z-vinylic secondary phosphine or with a prolonged reaction time, the 1,2-diphosphinoalkane.38
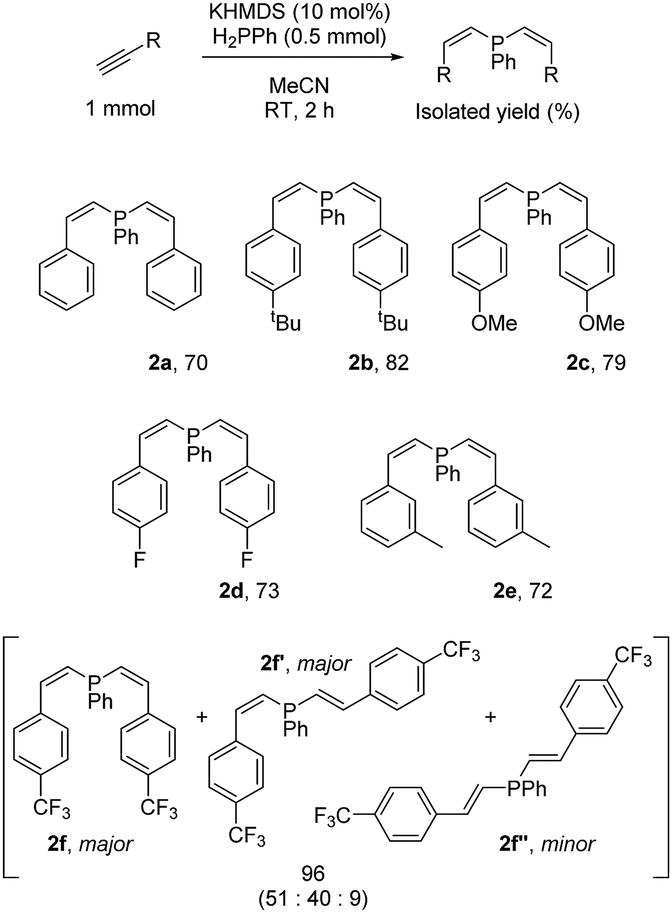 | ||
| Scheme 4 Substrate scope of double hydrophosphination of aryl acetylenes using H2PPh. | ||
In contrast to our chemistry with HPPh2 (vide supra), the KHMDS catalyzed transformation with H2PPh is limited to aryl acetylenes: when propiolates are employed complex mixtures form of what appears to be polymeric material. Phenyl acetylene and 4-tert-butylphenylacetylene react well, giving products 2a and 2b in high isolated yield. Crystals of 2a could be isolated and display carbon–carbon double bonds of 1.327(4) Å and 1.334(3) Å for C7–C8 and C15–C16 respectively and a C7–P1–C15 bond angle of 102.4(1)° (Fig. 2).
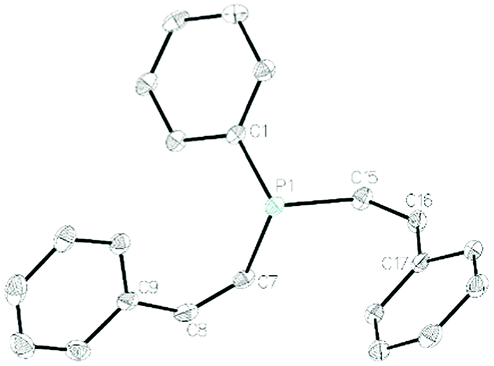 | ||
| Fig. 2 Molecular structure of 2a. Ellipsoids are represented at 30%. | ||
Phenyl rings substituted with electronically challenging groups selectively give the targeted products (2c and 2d). When a highly electron withdrawing 4-trifluoromethyl group is introduced to the aromatic ring a mixture of E- and Z-products form (2f, 2f′ and 2f′′). Mixtures of this type are not observed with the other aryl substrates. All products are obtained in near quantitative spectroscopic yield (>95%) and loss of product occurs due to the sensitivity of these vinyl phosphines.
In summary, we have presented a mild and rapid route to highly functionalized 1,1-diphosphines. NaOtBu, as an inexpensive and easy to handle base, can be used to transform simple propiolates into their 1,1-diphosphine congeners, with a high yield achieved even at 1 mol% loading. However, KHMDS is necessary for more challenging substrates. A change in phosphine reagent from diphenylphosphine to phenylphosphine results in a change in reactivity, allowing the selective formation of P,P-divinylphosphines. Both sets of products offer intriguing potential modes of reactivity: we envisage using some of our 1,1-diphosphines as bifunctional ligands for heterobimetallic complexes, whilst our divinylphosphines give facile access to monomers for the preparation of phosphorus-containing polymers. Both of these aspects are currently under investigation in our laboratory.
The University of Bath is thanked for a studentship (NTC). The EPSRC UK National Mass Spectrometry Facility at Swansea University is thanked for mass spectrometry analyses. This research was funded through an EPSRC Fellowship EP/P024254/1 (RLW). Crystallization of 1b was undertaken by Kimberley J. Gallagher.
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. C. J. Kamer and P. W. N. M. V. Leeuwen, Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, Wiley, 2012 Search PubMed.
- A. Börner, Phosphorus Ligands in Asymmetric Catalysis, Wiley-VCH, 2008 Search PubMed.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef.
- P. W. N. M. van Leeuwen, P. C. J. Kamer, J. N. H. Reek and P. Dierkes, Chem. Rev., 2000, 100, 2741–2770 CrossRef PubMed.
- P. Hofmann, C. Meier, W. Hiller, M. Heckel, J. Riede and M. U. Schmidt, J. Organomet. Chem., 1995, 490, 51–70 CrossRef.
- M. M. P. Grutters, C. Müller and D. Vogt, J. Am. Chem. Soc., 2006, 128, 7414–7415 CrossRef PubMed.
- G. M. Lee, C. M. Vogels, A. Decken and S. A. Westcott, Eur. J. Inorg. Chem., 2011, 2433–2438 CrossRef.
- A. B. Chaplin, J. F. Hooper, A. S. Weller and M. C. Willis, J. Am. Chem. Soc., 2012, 134, 4885–4897 CrossRef PubMed.
- I. Pernik, J. F. Hooper, A. B. Chaplin, A. S. Weller and M. C. Willis, ACS Catal., 2012, 2, 2779–2786 CrossRef.
- J. F. Hooper, R. D. Young, A. S. Weller and M. C. Willis, Chem. – Eur. J., 2013, 19, 3125–3130 CrossRef PubMed.
- A. Prades, M. Fernández, S. D. Pike, M. C. Willis and A. S. Weller, Angew. Chem., Int. Ed., 2015, 54, 8520–8524 CrossRef PubMed.
- S. K. Murphy, A. Bruch and V. M. Dong, Chem. Sci., 2015, 6, 174–180 RSC.
- R. S. Anju, B. Mondal, K. Saha, S. Panja, B. Varghese and S. Ghosh, Chem. – Eur. J., 2015, 21, 11393–11400 CrossRef PubMed.
- J. Lopez-Serrano, S. B. Duckett, J. P. Dunne, C. Godard and A. C. Whitwood, Dalton Trans., 2008, 4270–4281, 10.1039/B804162H.
- I. D. Gridnev, Y. Liu and T. Imamoto, ACS Catal., 2014, 4, 203–219 CrossRef.
- M. K. Cybulski, J. E. Nicholls, J. P. Lowe, M. F. Mahon and M. K. Whittlesey, Organometallics, 2017, 36, 2308–2316 CrossRef.
- A. Zanardo, R. A. Michelin, F. Pinna and G. Strukul, Inorg. Chem., 1989, 28, 1648–1653 CrossRef.
- M. M. Lok, L. K. Chung, S. W. Nga, N. S. Man, Z. Zhongyuan, L. Zhenyang and L. C. Po, Chem. – Eur. J., 2006, 12, 1004–1015 CrossRef PubMed.
- E. Pizzo, P. Sgarbossa, A. Scarso, R. A. Michelin and G. Strukul, Organometallics, 2006, 25, 3056–3062 CrossRef.
- M. Zablocka, N. Cénac, A. Igau, B. Donnadieu, J.-P. Majoral, A. Skowronska and P. Meunier, Organometallics, 1996, 15, 5436–5438 CrossRef.
- E. N. Tsvetkov, N. A. Bondarenko, I. G. Malakhova and M. I. Kabachnik, Synthesis, 1986, 198–208 CrossRef.
- M. T. Honaker, B. J. Sandefur, J. L. Hargett, A. L. McDaniel and R. N. Salvatore, Tetrahedron Lett., 2003, 44, 8373–8377 CrossRef.
- M. T. Honaker and R. N. Salvatore, Phosphorus, Sulfur Silicon Relat. Elem., 2004, 179, 277–283 CrossRef.
- C.-T. Yang, J. Han, J. Liu, Y. Li, F. Zhang, M. Gu, S. Hu and X. Wang, RSC Adv., 2017, 7, 24652–24656 RSC.
- C. Henri-Jean, V. David, M. Patrick and F. Alain, Eur. J. Org. Chem., 1999, 1561–1569 Search PubMed.
- F. Eisentrager, A. Gothlich, I. Gruber, H. Heiss, C. A. Kiener, C. Kruger, J. Ulrich Notheis, F. Rominger, G. Scherhag, M. Schultz, B. F. Straub, M. A. O. Volland and P. Hofmann, New J. Chem., 2003, 27, 540–550 RSC.
- A. Pews-Davtyan, X. Fang, R. Jackstell, A. Spannenberg, W. Baumann, R. Franke and M. Beller, Chem. – Asian J., 2014, 9, 1168–1174 CrossRef PubMed.
- A. M. Aguiar and T. G. Archibald, Tetrahedron Lett., 1966, 7, 5471–5475 CrossRef.
- K. Issleib, H. Böhme and C. Rockstroh, J. Prakt. Chem., 1970, 312, 571–577 CrossRef.
- G. Märkl and R. Potthast, Angew. Chem., Int. Ed., 1967, 6, 86 CrossRef.
- J. L. Bookham and D. M. Smithies, J. Organomet. Chem., 1999, 577, 305–315 CrossRef.
- Y. Zhang, L. Tang, Y. Ding, J.-H. Chua, Y. Li, M. Yuan and P.-H. Leung, Tetrahedron Lett., 2008, 49, 1762–1767 CrossRef.
- Reaction to afford 1d, 1i and 1j was undertaken at −78 °C to prevent unwanted side-products forming.
- See ESI†.
- H. Schmidbaur, G. Reber, A. Schier, F. E. Wagner and G. Müiller, Inorg. Chim. Acta, 1988, 147, 143–150 CrossRef.
- E. Kobayashi, T. Obata, S. Aoshima and J. Furukawa, Polym. J., 1993, 25, 1049 CrossRef.
- T. Miyaji, Z. Xi, M. Ogasawara, K. Nakajima and T. Takahashi, J. Org. Chem., 2007, 72, 8737–8740 CrossRef PubMed.
- C. A. Bange and R. Waterman, ACS Catal., 2016, 6, 6413–6416 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic methods, starting material and product analysis data and NMR spectra. CCDC 1849265–1849269 for 1b, 1f, 1i, 1k and 2a, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc05890c |
| This journal is © The Royal Society of Chemistry 2018 |