
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Further enhancement of the clickability of doubly sterically-hindered aryl azides by para-amino substitution†
Suguru
Yoshida
 a,
Junko
Tanaka
a,
Yoshitake
Nishiyama
a,
Yuki
Hazama
a,
Takeshi
Matsushita
b and
Takamitsu
Hosoya
*a
a,
Junko
Tanaka
a,
Yoshitake
Nishiyama
a,
Yuki
Hazama
a,
Takeshi
Matsushita
b and
Takamitsu
Hosoya
*a
aLaboratory of Chemical Bioscience, Institute of Biomaterials and Bioengineering, Tokyo Medical and Dental University (TMDU), 2-3-10 Kanda-Surugadai, Chiyoda-ku, Tokyo 101-0062, Japan. E-mail: thosoya.cb@tmd.ac.jp
bIchihara Research Center, JNC Petrochemical Corporation, 5-1 Goikaigan, Ichihara, Chiba 290-8551, Japan
First published on 15th October 2018
Abstract
Introduction of an amino group at the para position of doubly sterically-hindered aryl azides significantly enhances the reactivity with cyclooctynes. The distortability of the azido group is synergistically enhanced by the para-electron-donating group and two bulky ortho substituents, which increases the HOMO energy level and provoke the steric inhibition of resonance, respectively.
The click reaction,1,2 particularly the catalyst-free, strain-promoted azide–cycloalkyne cycloaddition (SPAAC),3,4 is one of the most reliable methods for conjugating molecules, having wide applications in broad research fields such as materials chemistry5 and chemical biology.6 To make the method more practical, a wide variety of useful cyclic alkynes with improved properties such as higher azidophilicity, lower hydrophobicity, and synthetic easiness have been developed.4 In contrast, little attention has been paid to increase the reactivity of the azide.7
In the course of our studies on azide chemistry,8 we found that 2,6-disubstituted phenyl azides show remarkably enhanced clickability in the reaction with strained alkynes such as Sondheimer diyne (1),9,10 despite the steric hindrance.8d From experimental and computational studies, we concluded that this seemingly contradictory phenomenon could be attributed to the increased distortability of the azido group elicited by the steric inhibition of resonance with the aromatic ring (Fig. 1). Compared to the electronic effects observed for the clickability of para-substituted phenyl azides (e.g., 2aversus2b), doubly sterically-hindered substrates such as 2,6-diisopropylphenyl azide (2c), in which the resonance is canceled to some extent, showed remarkably high reactivity. This finding enabled the facile preparation of trifunctional molecules by three sequential azido-type-selective triazole forming reactions using a triazido platform molecule bearing three different types of azido groups.8g In this study, with the aim of enhancing the selectivity of the click conjugation, the effect of para substituents on the clickability of 2,6-diisopropylphenyl azides was examined.
 | ||
| Fig. 1 Double-click reaction of Sondheimer diyne (1) with aryl azides 2.8dk: second-order rate constant. | ||
We firstly examined the reactivity of 2,6-diisopropylphenyl azides 2d–f bearing para-bromo, -iodo, and -pinacolatoboryl group, respectively, with diyne 1 (Table 1). Despite the different electronegativity of the three groups, little difference in clickability was observed between these azides (entries 2–4) and 2,6-diisopropylphenyl azide (2c) (entry 1).
We then turned our attention to the resonance effect that would be induced by the para substituent. For an efficient comparison, we prepared two types of 2,6-diisopropylphenyl azides 2g–k and 2l–o bearing a variety of para-aryl11 or -amino12 groups by the palladium-catalyzed cross-coupling reactions of 4-bromo-2,6-diisopropylaniline (4), followed by azidation of the resulting anilines via diazotization (Scheme 1). In addition, by means of the azido-type-selective cycloaddition8g of diazide 5 with acetylacetone catalyzed by potassium carbonate,13para-triazolyl-substituted 2,6-diisopropylphenyl azide 2p was prepared (Scheme 2).
 | ||
| Scheme 1 Preparation of azides 2g–o. | ||
 | ||
| Scheme 2 Preparation of azide 2p. | ||
The effect of para-aryl substituents on the clickability of 2,6-diisopropylphenyl azides was found quite limited in the reaction with diyne 1 (Table 2, entries 2–6). The reaction rates for azides 2g–j, bearing different aryl groups, i.e., phenyl, 2,6-xylyl, 4-(trifluoromethyl)phenyl, and 4-methoxyphenyl, respectively, were almost the same or smaller than that for azide 2c (entries 2–5), whereas 2k, which has an electron-rich 4-(dimethylamino)phenyl group, reacted slightly faster than 2c (entry 6).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | 2 | k (M−1 s−1) | k rel | Yield (%) | trans/cis |
| a From ref. 8d. b In acetonitrile. | ||||||
| 1 | H | 2c | 0.67a (0.37)b | 1 (1)b | 95a | 96/4a |
| 2 | Ph | 2g | 0.74 | 1.1 | Quant | 96/4 |
| 3 | 2,6-Me2C6H3 | 2h | 0.75 | 1.1 | 81 | 96/4 |
| 4 | 4-F3CC6H4 | 2i | 0.47 | 0.70 | Quant | 95/5 |
| 5 | 4-MeOC6H4 | 2j | 0.72 | 1.1 | Quant | 96/4 |
| 6 | 4-Me2NC6H4 | 2k | 0.83 | 1.2 | 88 | 96/4 |
| 7 |
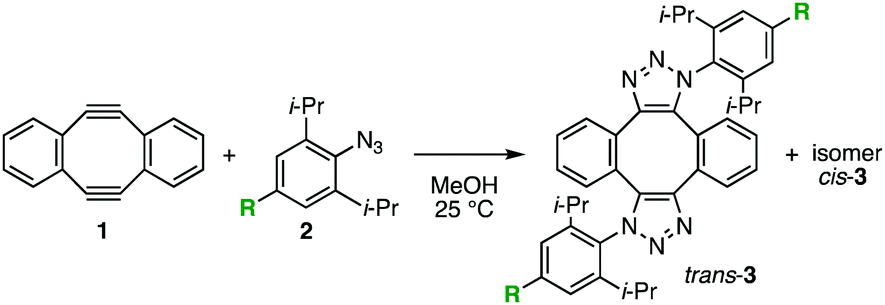
|
2l | 1.4 | 2.1 | Quant | 95/5 |
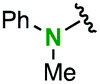
| 8 |

|
2m | 1.6 | 2.4 | 77 | 96/4 |
| 9 |
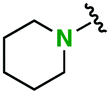
|
2n | 1.7 | 2.5 | Quant | 96/4 |
| 10 |

|
2o | 3.5 (2.1)b | 5.2 (5.8)b | Quant | 95/5 |
| 11 |
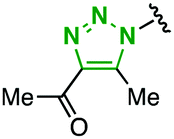
|
2p | 0.53 | 0.79 | Quant | 99/1 |
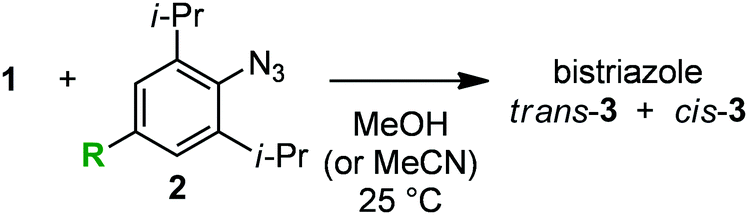
In marked contrast to the limited effect of the aryl, halogeno, and boryl groups, introduction of an amino group at the para position of 2,6-diisopropylphenyl azide significantly enhanced the clickability (Table 2, entries 7–11). For example, azides 2l–o bearing the para-amino groups reacted with 1 more than twice faster than 2c (entries 7–10). In particular, 2,6-diisopropyl-4-pyrrolidinophenyl azide (2o) showed the highest clickability, reacting more than five times faster than 2c (entry 10) and approximately 400 times faster than phenyl azide (2a). The enhanced clickability of azide 2o was also observed in the reaction with diyne 1 in acetonitrile, although the reaction rate decreased slightly compared to that in methanol (entry 10 versus entry 1). The reaction of azide 2p with 1 was rather slower than that of 2c, indicating that the triazolyl group was not effective in accelerating the reaction (entry 11).
Introduction of an amino group at the para position of phenyl azide also enhanced the clickablity of substrates without ortho substituents, as demonstrated in the reaction with diyne 1 (Table 3). For example, 4-morpholinophenyl azide (2q) showed slightly higher clickablity than 4-methoxyphenyl azide (2b) (entry 3 versus entry 2). The introduction of a morpholino group at the para position of phenyl azide elicited a 3.4-fold acceleration of the reaction (entry 3 versus entry 1). 4-Azidoaniline (2r), which has an unsubstituted amino group, also showed a comparable reactivity to that of the methoxy congener 2b (Table 3, entry 4 versus entry 2). Intriguingly, this accelerating effect was completely canceled by salt formation of the amino group of 4-azidoaniline, which resulted in the deceleration of the reaction (entry 5).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | 2 | k (M−1 s−1) | k rel | Yield (%) | trans/cis |
| a From ref. 8d. b In acetonitrile. | ||||||
| 1 | H | 2a | 8.8 × 10−3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (6.4 × 10−3)b a (6.4 × 10−3)b |
1 (1)b | 94a | 43/57a |
| 2 | MeO | 2b | 3.3 × 10−2a (1.8 × 10−2)b |
3.8 (2.7)b | 89a | 36/64a |
| 3 |
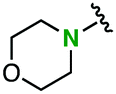
|
2q | (2.2 × 10−2)b | (3.4)b | Quant | 37/63 |
| 4 | H2N | 2r | 2.7 × 10−2 | 3.1 | Quant | 36/64 |
| 5 | ClH·H2N | 2s | 6.5 × 10−3 | 0.74 | Quant | 35/65 |

Frontier molecular orbital (FMO) analysis of azide 2c and para-amino-substituted 2,6-diisopropylphenyl azides 2m–o revealed a good correlation between the HOMO energy levels and the second-order rate constants (k) (Table 4). It was found that the reactivity of azides with diyne 1 increased with the rise of the HOMO energy. This result suggests that the electron-donating amino group at the para position reduced the energy gap between the HOMO of the aryl azide and the LUMO of diyne 1, which resulted in the enhancement of the cycloaddition. Indeed, azide 2o, which exhibited the highest HOMO energy due to the presence of the highly electron-donating pyrrolidino group, proved to be the most reactive among the azides examined.14
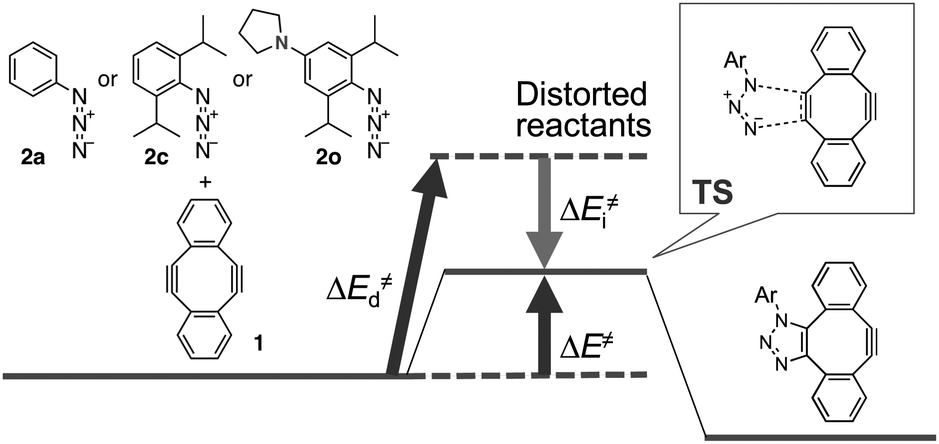
To gain further mechanistic insight into the enhanced clickability of para-amino-substituted phenyl azides, the transition state (TS) structure for the first cycloaddition of 1 with 2o was obtained by a density functional theory (DFT) (B3LYP/6-31G(d)) method15 and compared with those obtained for the reactions with 2a and 2c (Table 5). The activation energy for the reaction of 1 with 2o was lower than that with 2c, which was in good agreement with the experimental result. The corresponding distortion–interaction analysis16 indicated that, in accord with our previous report,8d the enhanced clickability of 2o mostly rested on the enhanced distortability of the azido group (2a: 17.4 kcal mol−1; 2c: 15.1 kcal mol−1; 2o: 13.8 kcal mol−1). We previously explained that the higher clickability of 2c compared to that of 2a was elicited by the steric inhibition of resonance: i.e., the two bulky ortho substituents of 2c forced the azido group to twist out of the plane of the benzene ring, which partially released the azido from the strong conjugation. In this case, however, the calculated rotational angle (θ) of the azido group from the aromatic plane for 2o (θ = 61.2°) was similar to that for 2c (θ = 58.2°) (Fig. 2), indicating that the steric inhibition of resonance alone cannot be invoked to rationalize the higher clickability of 2o compared to that of 2c. Taken together, the FMO and the distortion–interaction analyses suggest that the clickability of doubly sterically-hindered aryl azides bearing an amino group at the para position stemmed from the enhanced distortability of the azido group, which was synergistically provoked by the steric inhibition of resonance and the raised HOMO energy level.
|
|
||||||
|---|---|---|---|---|---|---|
| Reactants | 2a + 1b | 2c + 1b | 2o + 1 | |||
| a Distortion, interaction, and activation energies for the first cycloaddition obtained at the B3LYP/6-31G(d) are shown in kcal mol−1. b From ref. 8d. c Energy required to distort the geometry of each reactant to the transition state (TS). d Interaction energy between the distorted fragments at the TS. e Energy differences of each fragment between the optimized and the TS geometries. | ||||||
| Distorted reactantsc | +17.4 | +3.1 | +15.1 | +3.1 | +13.8 | +2.7 |
| Distortion energyc (ΔE‡d) | +20.4 | +17.8 | +16.5 | |||
| Interaction energyd (ΔE‡i) | −9.0 | −8.9 | −8.6 | |||
| Activation energye (ΔE‡) | +11.4 | +8.9 | +7.9 | |||
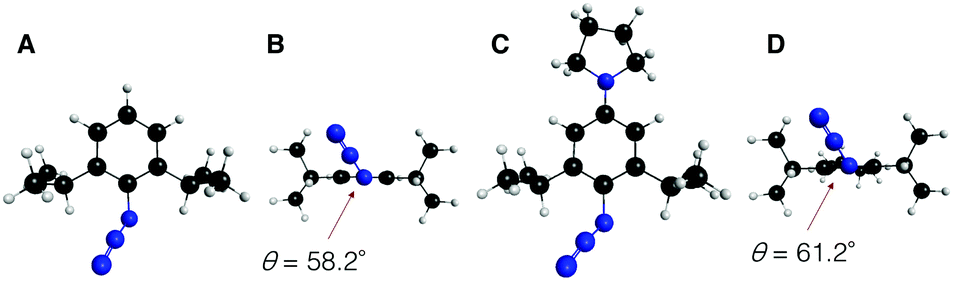 | ||
| Fig. 2 Global minima on the potential energy surface of aryl azides 2c and 2o obtained by DFT (B3LYP/6-31G(d)) method. (A) Overhead view of 2c. (B) Side view of 2c. (C) Overhead view of 2o. (D) Side view of 2o. θ indicates the rotational angle of the azido group from the aromatic plane. | ||
The clickability enhancement by para-amino substitution of sterically-hindered aryl azide was also observed in the SPAAC reactions using other cycloalkynes that are widely used for bioconjugation. The most reactive azide 2o in the reaction with diyne 1 also showed significantly higher reactivity in the reactions with a bicyclo[6.1.0]non-4-yne (BCN)4i derivative (k = 0.92 M−1 s−1) in MeOH than 2c (k = 0.20 M−1 s−1). The reaction of a 1:1 mixture of 2o and 2c with a dibenzoazacyclooctyne (DIBAC)4g derivative afforded 2o- and 2c-derived cycloadducts at a ratio of 3.3:1.17
The enhanced clickability of the aryl azide elicited by the para-amino group enabled performing a sequential double-click reaction using diazido compound 6 with excellent azido-type selectivity (Scheme 3). Thus, the reaction of 6 with the strained alkyne 7 took place exclusively at the sterically-hindered para-amino-substituted aryl azido group, despite the presence of another phenyl azido group with sufficient reactivity, and the subsequent copper-catalyzed azide–alkyne cycloaddition with terminal alkyne 8 afforded bistriazole 9 in high yield.
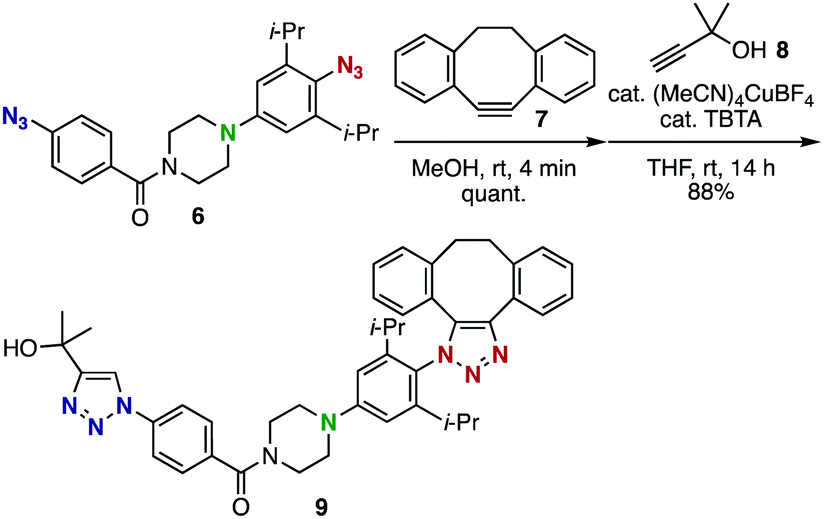 | ||
| Scheme 3 Azido-type selective double-click reaction of diazide 6. | ||
In summary, we have found that the introduction of an amino group at the para position of doubly sterically-hindered aryl azides significantly enhances their clickability in the reaction with strained alkynes. The azide is synergistically activated by the electron-donating para-amino group and two bulky ortho substituents, with the former increasing the HOMO energy level of the azide, thereby enhancing its ynophilicity, and the latter inhibiting the resonance between azido and phenyl groups, which enhances the distortability of the azido group. This finding allowed for the sequential double-click conjugation of a diazido compound with excellent efficiency, which would be useful for the preparation of multifunctionalized molecules using a multiazido platform molecule bearing a para-amino-2,6-disubstituted aryl azide unit. Further studies to develop more reactive azides for their application to the preparation of multifunctional molecules are ongoing.
This work was supported by AMED under Grant Numbers JP18am0101098 (Platform Project for Supporting Drug Discovery and Life Science Research, BINDS) and JP18am0301024 (the Basic Science and Platform Technology Program for Innovative Biological Medicine); JSPS KAKENHI Grant Numbers JP15H03118 and JP18H02104 (B; T. H.), JP16H01133 and JP18H04386 (Middle Molecular Strategy; T. H.), JP17H06414 (Organelle Zone; T. H.), JP26350971 (C; S. Y.), and JP17K13266 (Young Scientist B; Y. N.); the Cooperative Research Project of Research Center for Biomedical Engineering; and the Naito Foundation (S. Y.).
Conflicts of interest
There are no conflicts to declare.Notes and references
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS PubMed.
- (a) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef; (b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; (c) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952 CrossRef CAS PubMed; (d) J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302 RSC; (e) A. Mandoli, Molecules, 2016, 21, 1174 CrossRef PubMed.
- (a) E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974 CrossRef CAS PubMed; (b) M. F. Debets, C. W. J. van der Doelen, F. P. J. T. Rutjes and F. L. van Delft, ChemBioChem, 2010, 11, 1168 CrossRef CAS PubMed; (c) J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272 RSC; (d) J. Dommerholt, F. P. J. T. Rutjes and F. L. van Delft, Top. Curr. Chem., 2016, 374, 16 CrossRef PubMed.
- (a) G. Wittig and A. Krebs, Chem. Ber., 1961, 94, 3260 CrossRef CAS; (b) N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046 CrossRef CAS PubMed; (c) S. T. Laughlin, J. M. Baskin, S. L. Amacher and C. R. Bertozzi, Science, 2008, 320, 664 CrossRef CAS PubMed; (d) J. A. Codelli, J. M. Baskin, N. J. Agard and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 11486 CrossRef CAS PubMed; (e) X. Ning, J. Guo, M. A. Wolfert and G.-J. Boons, Angew. Chem., Int. Ed., 2008, 47, 2253 CrossRef CAS PubMed; (f) A. A. Poloukhtine, N. E. Mbua, M. A. Wolfert, G.-J. Boons and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 15769 CrossRef CAS PubMed; (g) M. F. Debets, S. S. van Berkel, S. Schoffelen, F. P. J. T. Rutjes, J. C. M. van Hest and F. L. van Delft, Chem. Commun., 2010, 46, 97 RSC; (h) J. C. Jewett, E. M. Sletten and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 3688 CrossRef CAS PubMed; (i) J. Dommerholt, S. Schmidt, R. Temming, L. J. A. Hendriks, F. P. J. T. Rutjes, J. C. M. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422 CrossRef CAS PubMed; (j) E. H. P. Leunissen, M. H. L. Meuleners, J. M. M. Verkade, J. Dommerholt, J. G. J. Hoenderop and F. L. van Delft, ChemBioChem, 2014, 15, 1446 CrossRef CAS PubMed; (k) S. Yoshida, Y. Hatakeyama, K. Johmoto, H. Uekusa and T. Hosoya, J. Am. Chem. Soc., 2014, 136, 13590 CrossRef CAS PubMed; (l) R. Ni, N. Mitsuda, T. Kashiwagi, K. Igawa and K. Tomooka, Angew. Chem., Int. Ed., 2015, 54, 1190 CrossRef CAS PubMed; (m) K. Kaneda, R. Naruse and S. Yamamoto, Org. Lett., 2017, 19, 1096 CrossRef CAS PubMed; (n) E. G. Burke, B. Gold, T. T. Hoang, R. T. Raines and J. M. Schomaker, J. Am. Chem. Soc., 2017, 139, 8029 CrossRef CAS PubMed.
- (a) J. Lahann, Click Chemistry for Biotechnology and Materials Science, John Wiley & Sons, West Sussex, 2009 CrossRef; (b) W. Xi, T. F. Scott, C. J. Kloxin and C. N. Bowman, Adv. Funct. Mater., 2014, 24, 2572 CrossRef CAS.
- (a) P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905 CrossRef CAS PubMed; (b) C. S. McKay and M. G. Finn, Chem. Biol., 2014, 21, 1075 CrossRef CAS PubMed.
- (a) S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188 CrossRef PubMed; (b) S. Bräse and K. Banert, Organic Azides: Syntheses and Applications, John Wiley & Sons, West Sussex, 2010 Search PubMed; (c) Z. Yuan, G.-C. Kuang, R. J. Clark and L. Zhu, Org. Lett., 2012, 14, 2590 CrossRef CAS PubMed; (d) J. Dommerholt, O. van Rooijen, A. Borrmann, C. F. Guerra, F. M. Bickelhaupt and F. L. van Delft, Nat. Commun., 2014, 5, 5378 CrossRef CAS PubMed; (e) S. Xie, S. A. Lopez, O. Ramström, M. Yan and K. N. Houk, J. Am. Chem. Soc., 2015, 137, 2958 CrossRef CAS PubMed; (f) N. Münster, P. Nikodemiak and U. Koert, Org. Lett., 2016, 18, 4296 CrossRef PubMed; (g) K. Banert, Synthesis, 2016, 48, 2361 CrossRef CAS; (h) D. Huang and G. Yan, Adv. Synth. Catal., 2017, 359, 1600 CrossRef CAS.
- (a) T. Hosoya, T. Hiramatsu, T. Ikemoto, M. Nakanishi, H. Aoyama, A. Hosoya, T. Iwata, K. Maruyama, M. Endo and M. Suzuki, Org. Biomol. Chem., 2004, 2, 637 RSC; (b) T. Hosoya, T. Hiramatsu, T. Ikemoto, H. Aoyama, T. Ohmae, M. Endo and M. Suzuki, Bioorg. Med. Chem. Lett., 2005, 15, 1289 CrossRef CAS PubMed; (c) T. Hosoya, A. Inoue, T. Hiramatsu, H. Aoyama, T. Ikemoto and M. Suzuki, Bioorg. Med. Chem., 2009, 17, 2490 CrossRef CAS PubMed; (d) S. Yoshida, A. Shiraishi, K. Kanno, T. Matsushita, K. Johmoto, H. Uekusa and T. Hosoya, Sci. Rep., 2011, 1, 82 CrossRef PubMed; (e) S. Yoshida, Y. Misawa and T. Hosoya, Eur. J. Org. Chem., 2014, 3991 CrossRef CAS; (f) T. Meguro, S. Yoshida and T. Hosoya, Chem. Lett., 2017, 46, 1137 CrossRef CAS; (g) S. Yoshida, K. Kanno, I. Kii, Y. Misawa, M. Hagiwara and T. Hosoya, Chem. Commun., 2018, 54, 3705 RSC; (h) T. Meguro, N. Terashima, H. Ito, Y. Koike, I. Kii, S. Yoshida and T. Hosoya, Chem. Commun., 2018, 54, 7904 RSC; (i) T. Meguro, S. Yoshida, K. Igawa, K. Tomooka and T. Hosoya, Org. Lett., 2018, 20, 4126 CrossRef CAS PubMed.
- (a) H. N. C. Wong, P. J. Garratt and F. Sondheimer, J. Am. Chem. Soc., 1974, 96, 5604 CrossRef CAS; (b) A. Orita, D. Hasegawa, T. Nakano and J. Otera, Chem. – Eur. J., 2002, 8, 2000 CrossRef CAS PubMed; (c) F. Xu, L. Peng, K. Shinohara, T. Morita, S. Yoshida, T. Hosoya, A. Orita and J. Otera, J. Org. Chem., 2014, 79, 11592 CrossRef CAS PubMed.
- I. Kii, A. Shiraishi, T. Hiramatsu, T. Matsushita, H. Uekusa, S. Yoshida, M. Yamamoto, A. Kudo, M. Hagiwara and T. Hosoya, Org. Biomol. Chem., 2010, 8, 4051 RSC.
- A. S. Guram, A. O. King, J. G. Allen, X. Wang, L. B. Schenkel, J. Chan, E. E. Bunel, M. M. Faul, R. D. Larsen, M. J. Martinelli and P. J. Reider, Org. Lett., 2006, 8, 1787 CrossRef CAS PubMed.
- X.-X. Zhang and S. L. Buchwald, J. Org. Chem., 2000, 65, 8027 CrossRef CAS PubMed.
- E. P. J. Ng, Y.-F. Wang, B. W.-Q. Hui, G. Lapointe and S. Chiba, Tetrahedron, 2011, 67, 7728 CrossRef CAS.
- E. Kolehmainen, R. Gawinecki and B. Osmiałowski, in The Chemistry of Anilines Part 1, ed. Z. Rappoport, John Wiley & Sons, West Sussex, 2007, pp. 347–371 Search PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery Jr., J. Comput. Chem., 1993, 14, 1347 CrossRef CAS.
- (a) D. H. Ess and K. N. Houk, J. Am. Chem. Soc., 2008, 130, 10187 CrossRef CAS PubMed; (b) F. Schoenebeck, D. H. Ess, G. O. Jones and K. N. Houk, J. Am. Chem. Soc., 2009, 131, 8121 CrossRef CAS PubMed; (c) F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070 CrossRef CAS PubMed.
- See the ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization for new compounds including NMR spectra. See DOI: 10.1039/c8cc05791e |
| This journal is © The Royal Society of Chemistry 2018 |