
Cyanomethyl anion transfer reagents for diastereoselective Corey–Chaykovsky cyclopropanation reactions†
Renè
Hommelsheim
 a,
Katharina J.
Hock
a,
Christian
Schumacher
a,
Mohanad A.
Hussein
b,
Thanh V.
Nguyen
*b and
Rene M.
Koenigs
*a
a,
Katharina J.
Hock
a,
Christian
Schumacher
a,
Mohanad A.
Hussein
b,
Thanh V.
Nguyen
*b and
Rene M.
Koenigs
*a
aRWTH Aachen University, Institute of Organic Chemistry, Landoltweg 1, 52074 Aachen, Germany. E-mail: rene.koenigs@rwth-aachen.de; Web: http://www.koenigslab.rwth-aachen.de
bUniversity of New South Wales, School of Chemistry, Sydney, Australia. E-mail: t.v.nguyen@unsw.edu.au; Web: https://tvnguyengroup.wordpress.com
First published on 20th September 2018
Abstract
A readily available and bench-stable cyanomethyl sulfonium salt was used in highly diastereoselective Corey–Chaykovsky cyclopropanation reactions of electron-poor olefins. This efficient method provides a rapid route to access densely functionalized cyclopropyl nitriles.
Cyclopropanes are a fascinating class of small molecules and possess unique properties that are reasoned by the conformational rigidity and distinctive spatial arrangement of substituents in this smallest-possible carbocycle.1 Cyclopropyl nitriles are ubiquitous in pharmaceutical research, for example as Cathepsin C inhibitors (1, Fig. 1) or as NMDA receptor modulators (2).3 They are also valuable precursors for the synthesis of cyclopropyl methylamines,2 as the reduction of the nitrile group readily furnishes cyclopropyl methylamines. These compounds in turn are privileged scaffolds in many biologically active compounds (3–5) and find applications in marketed drugs, e.g. Tasimelteon (4) or Levomilnacipran (5).3,4 The latter was approved in 2013 and is used for the treatment of depression (Fig. 1).
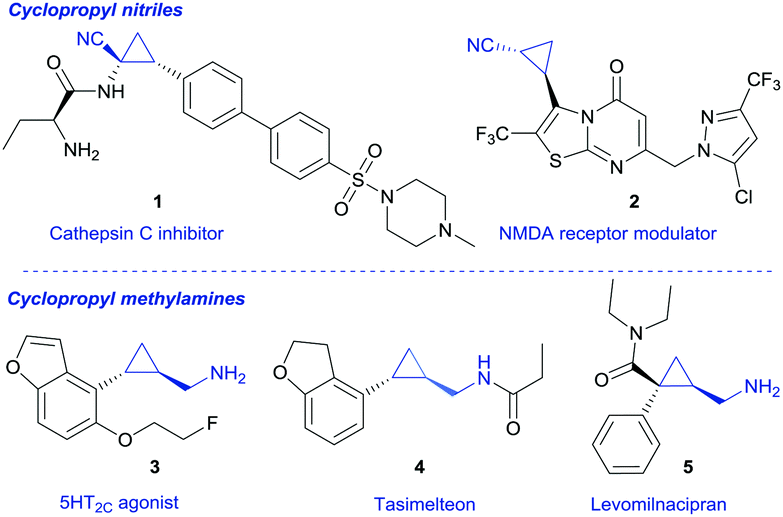 | ||
| Fig. 1 Applications of cyclopropyl nitriles and cyclopropyl methylamines. | ||
Despite tremendous research in the synthesis of ester or trifluoromethyl-substituted cyclopropane ring systems,1,5 synthetic methods of nitrile-substituted cyclopropanes are still scarce.6 Even today, the introduction of a “cyanomethylene” group remains a challenge to organic synthetic methodology7 and only normally proceeds either by transfer of a cyanomethyl carbene6a or cyanomethyl radical.6b From a synthetic perspective, traditional approaches are limited with respect to the use of either expensive catalysts or hazardous reagents. From a diversity point of view, they only allow the synthesis of trans-substituted nitrile-cyclopropanes with moderate diastereoselectivity. It is thus not surprising that current applications of nitrile cyclopropanes are almost exclusively on trans-substituted cyclopropanes.
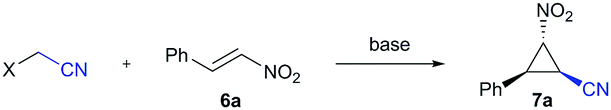
We therefore became intrigued in exploring alternative reagents for the introduction of a cyanomethylene group6,8 and envisaged a Michael initiated ring closing reaction using a cyanomethylene anion transfer reagent (Scheme 1). In previous studies,9 it was already demonstrated that halogen-substituted acetonitrile provides an access to nitrile-cyclopropanes though with strict limitations to substrate scope, yield and stereoselectivity.
 | ||
| Scheme 1 Cyanomethyl anion transfer. | ||
Inspired by these findings, we set out to explore the cyclopropanation reaction between nitro styrene, as a model substrate for electron-deficient olefins to prepare functionalized cyclopropanes, and halo-acetonitrile in the presence of different organic and inorganic bases (Table 1). Unfortunately, only the rapid decomposition of the halo-acetonitrile was observed under these basic reaction conditions. We subsequently evaluated different types of nucleophilic additives that could potentially form an intermediate onium salt, which is more acidic and should require less basic reaction conditions to proceed (Table 1, entries 3–9). Of all additives tested, only dimethyl sulfide gave some positive results and cyclopropane 7a was isolated in 10% yield. The screening of different reaction conditions did not lead to an improvement of this transformation.
|
|
||||
|---|---|---|---|---|
| #a | X | Base | Additives | Yield (7a) |
| a Reaction conditions: 6a (0.2 mmol), 2 eq. halo acetonitrile and 1.5 eq. of base were stirred in 2 mL of DCM for 12 h at rt. Yields refer to isolated products as single diastereoisomer. | ||||
| 1 | Br, Cl | NEt3, DBU, TBAT, TBAF | — | No rct. |
| 2 | Br, Cl | Li2CO3, Na2CO3, K2CO3, Cs2CO3, CsF, NaOEt, KOH | — | No rct. |
| 3 | Br | K2CO3 | Me2S | 10% |
| 4 | Br | Na2CO3 | Me2S | Traces |
| 5 | Br | CsF | Me2S | No rct. |
| 6 | Br | NEt3 | Me2S | No rct. |
| 7 | Br | K2CO3 | PhSMe | No rct. |
| 8 | Br | K2CO3 | Ph2S | No rct. |
| 9 | Br | K2CO3 | DMAP | No rct. |
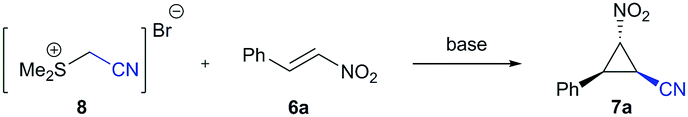
We thus decided to exchange the halogen moiety by a neutral leaving group and turned our attention to the preformed sulfonium salt 8 (Table 2) for Corey–Chaykovsky cyclopropanation reactions as a substitute of bromo acetonitrile.10 In 1966, Baizer described the synthesis of a cyanomethyl sulfonium salt for the first time,11a which is being used today as a versatile coupling reagent in peptide synthesis.11,12 From a synthetic perspective, sulfonium salts are much less explored, e.g. in carbonyl homologation reactions11b,d or in the synthesis of pyridine-2H-ones and isatines.11c,d Jeckel and Gosselck reported the reaction with malonic acid derivatives in cyclopropanation reactions, though only two single examples are provided.11f The deprotonation of a cyanomethyl sulfonium salt provides a sulfur ylide, whose nucleophilic and kinetic properties were well studied by Mayr and co-workers.13
|
|
||||
|---|---|---|---|---|
| #a | Base | Solvent | Concentration (M) | Yield (%) (7a) |
| a Reaction conditions: 6a (0.2 mmol), 2 eq. 8 and 1.5 eq. of Na2CO3 were stirred in 2 mL of DCM for 12 h at rt. Yields refer to isolated products as single diastereoisomer. b Reaction with Z-nitro styrene. | ||||
| 1 | Li2CO3 | DCM | 0.2 | Traces |
| 2 | Na2CO3 | DCM | 0.2 | 76 |
| 3 | K2CO3 | DCM | 0.2 | 63 |
| 4 | Cs2CO3 | DCM | 0.2 | 60 |
| 5 | NEt3 | DCM | 0.2 | 64 |
| 6 | CsF | DCM | 0.2 | 73 |
| 7 | TBAT | DCM | 0.2 | 43 |
| 8 | Na2CO3 | EtOAc | 0.2 | 53 |
| 9 | Na2CO3 | THF | 0.2 | 55 |
| 10 | Na2CO3 | 1,4-Dioxane | 0.2 | 72 |
| 11 | Na2CO3 | CHCl3 | 0.2 | 62 |
| 12 | Na2CO3 | DCM | 0.8 | 60 |
| 13 | Na 2 CO 3 | DCM | 0.1 | 93 |
| 14b | Na2CO3 | DCM | 0.1 | 43 |
We examined different bases and solvents for the cyclopropanation reaction (Table 2) and in all cases we could isolate the desired cyclopropane as a single diastereoisomer, which was identified as the trans-isomer 7a (referring to the relative stereochemistry of the nitro and nitrile moiety) as determined by NOE spectroscopy.14 The aryl group is arranged in cis stereochemistry to the nitrile group. Only in the case of TBAT (tetrabutylammonium triphenyl difluorosilicate, entry 7) we could observe a trace amount of a second isomer, which was identified as the all-cis isomer as shown by NOE experiments. The concentration had a significant effect on the yield (entries 2, 12 and 13) and the combination of sodium carbonate, DCM solvent and a concentration of 0.1 M proved to be the best conditions for the Corey–Chaykovsky cyclopropanation reaction, which proceeded in excellent yield and diastereoselectivity. The cis stereochemistry of the nitrile group and the aromatic ring is remarkable and highlights differences in reactivity and complementarity of the carbene5a and radical reactivity5bversus the Corey–Chaykovsky reactivity8a,10 as now both diastereoisomers can be accessed with a high degree of stereo control. We also investigated Z-nitro styrene and obtained a single product, which turned out to be the same diastereoisomer as for the E-nitro olefin, albeit in reduced yield.
In further experiments, we studied different nitro olefins to evaluate the applicability of this transformation. Different electron-withdrawing and electron-donating substituents in ortho-, meta- or para-position were well-tolerated and the desired cyclopropanes were isolated as a single diastereoisomer with good to high yields (Scheme 2, entries 7b–7l). Similarly, heterocycles, such as thiophene (7p), furane (7o) or pyridine (7n) and other carbocycles (7m) were tolerated and the corresponding nitro olefins reacted smoothly to afford the cyclopropanation products. Notably, an aliphatic substrate was converted to the desired cyclopropane (7q) in good isolated yield. In the case of β-methyl-trans-β-nitro styrene, the cyclopropane was obtained only in low yield, albeit as a single diastereoisomer (7r). On the other hand, α-methyl-trans-β-nitro styrene did not provide the desired cyclopropanation product; both these results can be attributed to steric hindrance of the methyl group at the nitro olefin. In a scale-up of this cyclopropanation reaction we could demonstrate that the desired cyclopropane 7a can be obtained on gram-scale (1.04 g 7a, 82%, 6.7 mmol reaction scale).
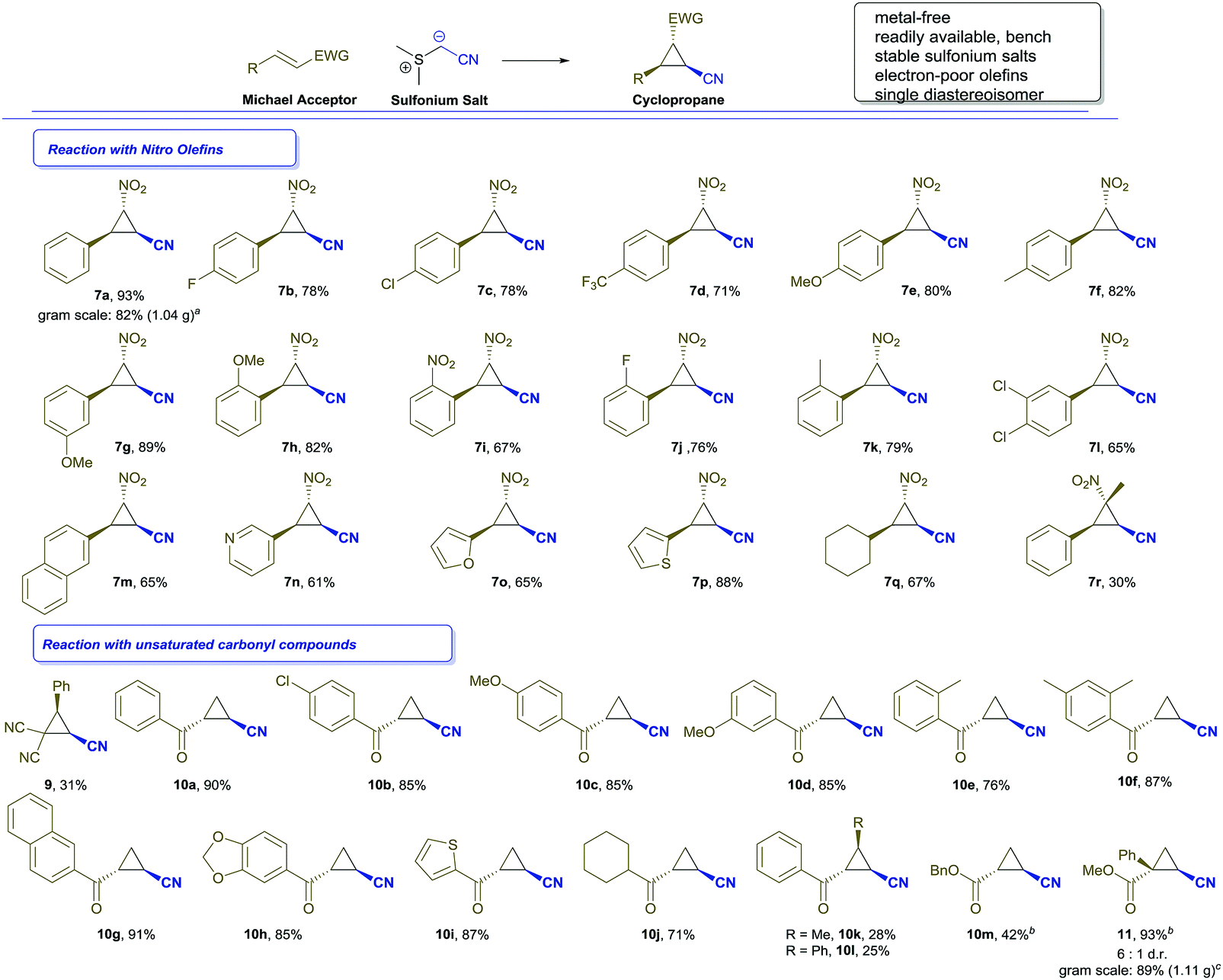 | ||
Scheme 2 Substrate scope of nitro olefins and unsaturated carbonyl compounds. Reaction conditions: Michael acceptor (0.2 mmol), 2 eq. 8 and 1.5 eq. of Na2CO3 were stirred in 2 mL of DCM for 12 h at rt. Yields refer to isolated products as single diastereoisomer; a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6.7 mmol scale; busing 1.75 eq. Cs2CO3 in 2 mL 1,4-dioxane; c6.2 mmol scale. 6.7 mmol scale; busing 1.75 eq. Cs2CO3 in 2 mL 1,4-dioxane; c6.2 mmol scale. | ||
To further expand the scope and applicability of this cyclopropanation reaction, we examined different unsaturated carbonyl compounds. Interestingly, typical Michael acceptors, such as cinnamic aldehyde, nitrile or ethyl ester did not provide the desired reaction products. Only in the case of benzylidene malodinitrile the cyclopropanation product (9) could be isolated, though with poor yield. We next turned our attention to terminal α,β-unsaturated ketones. In our model reaction, we were delighted to observe that the desired cyclopropane 10a could be isolated as a single diastereoisomer, irrespective of solvent and base, which is also the trans-isomer as shown by NOE spectroscopy.14 The substitution pattern on the aromatic rings of other analogous substrates (Scheme 2, entry 10b–10f) had little influence on the cyclopropanation reaction. Similarly, sulfur-containing heterocycles (10i), carbocycles (10g) and aliphatic substrates (10j) reacted smoothly under the present conditions. β-Substituted unsaturated ketones or benzyl acrylate gave significantly diminished yields (10k–m), which might be attributed to steric hindrance due to the substituent in the β-position. In all cases the trans configured product was obtained exclusively as a single diastereoisomer.14
Finally, we decided to study applications of this protocol in the synthesis of a key precursor of Levomilnacipran (5). For this purpose, we investigated the reaction of 2-phenyl acrylic acid ester with our sulfonium salt. To our delight, this ester reacted smoothly to form the desired cyclopropane 11 in excellent yield, albeit in moderate diastereoselectivity. Unfortunately, even after extensive screening of solvents and bases the diastereoselectivity of this transformation could only be improved to a 6:1 ratio.14 This can be rationalized by the lack of steric discrimination between the ester and phenyl groups. In a gram-scale experiment we were able to demonstrate the scalability of this transformation (1.11 g 11, 89%, 6.2 mmol reaction scale). The stereochemistry of the major product was confirmed to be trans by comparison with literature data.15
The stereochemistry of this cyclopropanation reaction is consistent with previous observations8a in Corey–Chaykovsky cyclopropanation reactions and can be rationalized by the mechanism depicted in Scheme 3a. We believe the cyclopropanation of Z-nitro styrene involved a re-orientation process when the nitro group on the carbanion intermediate rotated to avoid steric clash with the cyano group (see step 2 of mechanism depicted in Scheme 3b) to prepare for the SN2 cyclopropanation, hence forming the same product as the E-isomer.
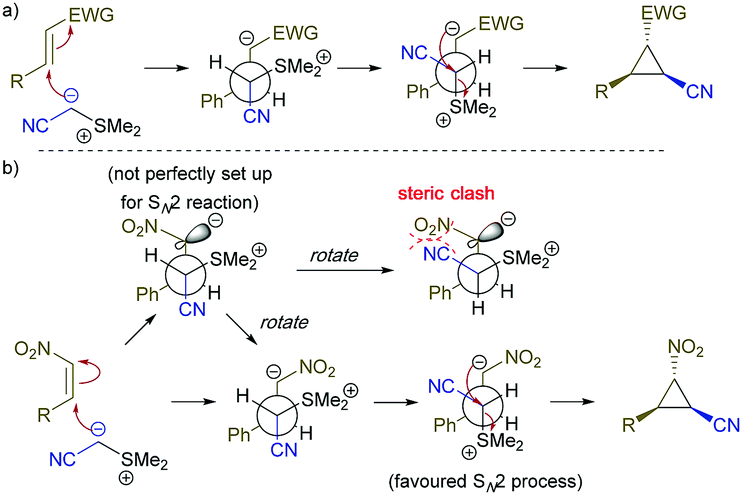 | ||
| Scheme 3 Proposed mechanism for the for the cyclopropanation reaction, (a) reaction with electron poor olefins, (b) reaction with Z-nitro olefin. | ||
In summary, we have developed a novel and robust protocol for the highly diastereoselective synthesis of nitrile-substituted cyclopropanes. We could demonstrate applications of a (cyanomethyl) dimethyl sulfonium salt in cyclopropanation reactions of electron-poor olefins and showcase their potential as a surrogate for hazardous diazoacetonitrile carbene precursor. This protocol features a broad functional group tolerance, including a diverse set of heterocyclic- and aliphatic substituted electron-poor olefins. Moreover, this approach can be readily applied to the synthesis of a key synthetic precursor of Levomilnacipran.
RMK thanks for support from the Excellence Initiative of the German federal and state governments and the Fonds der Chemischen Industrie (Sachkostenbeihilfe). The authors thank the Australian Research Council (grant DE150100517) for financial support. MAH thanks the Iraqi SCED for sponsoring his PhD scholarship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) D. Y. K. Chen, R. H. Pouwer and J.-A. Richard, Chem. Soc. Rev., 2012, 41, 4631 RSC; (b) T. F. Schneider, J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2014, 53, 5504 CrossRef PubMed; (c) H. U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151 CrossRef PubMed; (d) A. de Meijere and S. I. Kozhushkov, Sci. Synth., 2009, 48, 477 Search PubMed; (e) L. A. Wessjohann, W. Brandt and T. Thiemann, Chem. Rev., 2003, 103, 1625 CrossRef PubMed; (f) L. Mertens and R. M. Koenigs, Org. Biomol. Chem., 2016, 14, 10547 RSC; (g) O. O. Grygorenko, O. S. Artamonov, I. V. Komarov and P. K. Mykhailiuk, Tetrahedron, 2011, 67, 803 CrossRef.
- (a) A. C. Flick, H. X. Ding, C. A. Leveretti, R. E. Kyne, K. K.-C. Liu, S. J. Fink and C. J. O’Donnell, Bioorg. Med. Chem., 2016, 24, 1937 CrossRef PubMed; (b) J. D. Catt, G. Johnson, D. J. Keavy, R. J. Mattson, M. F. Parker, K. S. Takaki and J. P. Yevich, US pat., US5856529, Bristol-Myers Squibb, 1999.
- (a) J. Pedersen and C. Lauritzen, WO2012130299, Prozymex A/S, 2012; (b) M. Volgraf, B. D. Sellers, Y. Jiang, G. Wu, C. Q. Ly, E. Villemure, R. M. Pastor, P.-W. Yuen, A. Lu, X. Luo, M. Liu, S. Zhang, L. Sun, Y. Fu, P. J. Lupardus, H. J. A. Wallweber, B. M. Liederer, G. Deshmukh, E. Plise, S. Tay, P. Reynen, J. Herrington, A. Gustafson, Y. Liu, A. Dirksen, M. G. A. Dietz, Y. Liu, T.-M. Wang, J. E. Hanson, D. Hackos, K. Scearce-Levie and J. B. Schwarz, J. Med. Chem., 2016, 59, 2760 CrossRef PubMed; (c) A. P. S. Narula, E. M. Arruda, A. J. Janczuk and F. T. Schiet, US pat., US20060287204, International Flavors and Fragrances Inc., 2006; (d) J. Cheng, J. D. McCorvy, P. M. Giguere, H. Zhu, T. Kenakin, B. L. Roth and A. P. Kozikowski, J. Med. Chem., 2016, 59, 9866 CrossRef PubMed.
- (a) P. M. Farina, R. I. Rodriguez Curiel, S. Maiorana, A. Bianchi, F. Colombo and G. Timpano, WO2015092502, Laboratorio Chimico Internazionale, 2015; (b) S. Shuto, S. Ono, Y. Hase, N. Kamiyama and A. Matsuda, Tetrahedron Lett., 1996, 37, 641 CrossRef; (c) M. Doyle and W. Hu, Adv. Synth. Catal., 2001, 343, 299 CrossRef.
- Selected references on fluorinated cyclopropanes: (a) Y. Duan, B. Zhou, J.-H. Lin and J.-C. Xiao, Chem. Commun., 2015, 51, 13127 RSC; (b) K. J. Hock, L. Mertens and R. M. Koenigs, Chem. Commun., 2016, 52, 13783 RSC; (c) Y. Duan, J.-H. Lin, J.-C. Xiao and Y.-C. Gu, Org. Lett., 2016, 18, 2471 CrossRef PubMed; (d) B. Morandi and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 39, 938 CrossRef PubMed.
- (a) K. J. Hock, R. Spitzner and R. M. Koenigs, Green Chem., 2017, 19, 2118 RSC; (b) K. Thommes, G. Kiefer, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2009, 48, 8115 CrossRef PubMed.
- H. Wang, Y. Shao, H. Zheng, H. Wang, J. Cheng and X. Wan, Chem. – Eur. J., 2015, 21, 18333 CrossRef PubMed.
- (a) K. J. Hock, R. Hommelsheim, L. Mertens, J. Ho, T. Vinh Nguyen and R. M. Koenigs, J. Org. Chem., 2017, 82, 8220 CrossRef PubMed; (b) U. P. N. Tran, K. J. Hock, C. P. Gordon, R. M. Koenigs and T. V. Nguyen, Chem. Commun., 2017, 53, 4950 RSC; (c) K. J. Hock, L. Mertens, R. Hommelsheim, R. Spitzner and R. M. Koenigs, Chem. Commun., 2017, 53, 6577 RSC; (d) K. J. Hock and R. M. Koenigs, Angew. Chem., Int. Ed., 2017, 56, 13566 CrossRef PubMed.
- (a) M. Keita, R. De Bona, M. Dos Santos, O. Lequin, S. Ongeri, T. Milcent and B. Crousse, Tetrahedron, 2013, 69, 3308 CrossRef; (b) M. M. Kayser, J. Salvador, P. Morand and H. G. Krishnamurty, Can. J. Chem., 1982, 60, 1199 CrossRef.
- (a) E. J. Corey and M. Chaykovsky, J. Am. Chem. Soc., 1962, 84, 3782 CrossRef; (b) E. J. Corey and M. Chaykovsky, J. Am. Chem. Soc., 1965, 87, 1353 CrossRef; (c) V. Aggarwal and J. Richardson, Sci. Synth., 2004, 27, 21 Search PubMed.
- (a) M. M. Baizer, J. Org. Chem., 1966, 31, 3847 CrossRef; (b) Y. Hattori, K. Kobayashi, A. Deguchi, Y. Nohara, T. Akiyama, K. Teruya, A. Sanjoh, A. Nakagawa, E. Yamashita and K. Akaji, Bioorg. Med. Chem. Lett., 2015, 23, 5626 CrossRef PubMed; (c) C. T. Lollar, K. M. Krenek, K. J. Bruemmer and A. R. Lippert, Org. Biomol. Chem., 2014, 12, 406 RSC; (d) Q. Zhang, X. Liu, X. Xin, R. Zhang, Y. Liang and D. Dong, Chem. Commun., 2014, 50, 15378 RSC; (e) M. M. Ahire, M. B. Thoke and S. B. Mhaske, Org. Lett., 2018, 20, 848 CrossRef PubMed; (f) D. Jeckel and J. Gosselck, Tetrahedron Lett., 1972, 13, 2101 CrossRef; (g) B. Trupp, D.-R. Handreck, H.-P. Böhm, L. Knothe, H. Fritz and H. Prinzbach, Chem. Ber., 1991, 124, 1757 CrossRef.
- (a) L. Ju, A. R. Lippert and J. W. Bode, J. Am. Chem. Soc., 2008, 130, 4253 CrossRef PubMed; (b) V. R. Pattabiraman, A. O. Ogunkoya and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 5114 CrossRef PubMed; (c) T. Fukuzumi, L. Ju and J. W. Bode, Org. Biomol. Chem., 2012, 10, 5837 RSC.
- R. Appel and H. Mayr, Chem. – Eur. J., 2010, 16, 8610 CrossRef PubMed.
- For details, please see ESI†.
- M. Basato, C. Tubaro, A. Biffis, M. Bonato, G. Buscemi, F. Lighezzolo, P. Lunardi, C. Vianini, F. Benetollo and A. del Zotto, Chem. – Eur. J., 2009, 15, 1516 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc05602a |
| This journal is © The Royal Society of Chemistry 2018 |