
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The semiquinone radical anion of 1,10-phenanthroline-5,6-dione: synthesis and rare earth coordination chemistry†
James R.
Hickson
ab,
Samuel J.
Horsewill
a,
Jake
McGuire
 a,
Claire
Wilson
a,
Stephen
Sproules
*a and
Joy H.
Farnaby
*a
a,
Claire
Wilson
a,
Stephen
Sproules
*a and
Joy H.
Farnaby
*a
aSchool of Chemistry, WestCHEM, University of Glasgow, Glasgow, UK. E-mail: joy.farnaby@glasgow.ac.uk; Stephen.sproules@glasgow.ac.uk
bDepartment of Chemistry, Imperial College London, South Kensington, London, UK
First published on 18th September 2018
Abstract
Reduction of 1,10-phenanthroline-5,6-dione (pd) with CoCpR2 resulted in the first molecular compounds of the pd˙− semi-quinone radical anion, [CoCpR2]+[pd]˙− (R = H, (1); R = Me4, (2)). Furthermore compounds 1 and 2 were reacted with [Y(hfac)3(thf)2] (hfac = 1,1,1-5,5,5-hexafluoroacetylacetonate) to synthesise the rare earth-transition metal heterometallic compounds, [CoCpR2]+[Y(hfac)3(N,N′-pd)]˙− (R = H, (3); R = Me4, (4)).
Electron transfer reactions are fundamental to chemical processes. Stable organic and organometallic radicals are widely utilised, particularly in small molecule activation and catalysis.1 Redox-active ligands can act as electron reservoirs to achieve two electron processes, such as oxidative addition or reductive elimination, that would otherwise be inaccessible. This is a successful strategy in transition metal chemistry for the replacement of precious metals with earth abundant metals2 and has also been demonstrated for the f-elements.3,4
The most common use of a f-element radical is the use of samarium diiodide in organic chemistry. However, organometallic complexes of the f-elements have proved particularly suited for the reductive activation and stabilisation in well-defined molecular complexes, of some of the most unusual and biologically relevant radical species e.g. CO˙−, CO2˙− and NO˙−.5–7 The number of examples of f-elements complexed to redox-active ligands or radical anions is still modest but this is a growing area of research activity; for magnetism, multi-electron transfer chemistry and functional materials.4,8 Heterobimetallic complexes remain a particular synthetic challenge. In order to realise mixed-metal complexes, a bridging ligand with different coordination sites is needed so that metal ions can be selected and installed step-wise to acquire the desired properties. However, the bridging ligands used in f-element chemistry have identical binding sites that lead near exclusively to homobimetallic complexes.
Phenanthroline dione (pd) is a commercially available yet highly underutilised redox-active ligand. It constitutes a three-membered electron transfer series, where the neutral form can be sequentially, one-electron reduced to give the monoanionic and dianionic forms (Scheme 1). The terminal members are diamagnetic, whereas the monoanionic form is a radical with an S = 1/2 ground state. The most prized features of this ligand are its two binding sites. In the dianionic form of the ligand (right, Scheme 1), the N,N′-pocket is a π-accepting diimine unit analogous to 2,2′-bipyridine, and distinct from the O,O′-pocket which is a π-donating diolate, akin to a catecholate.
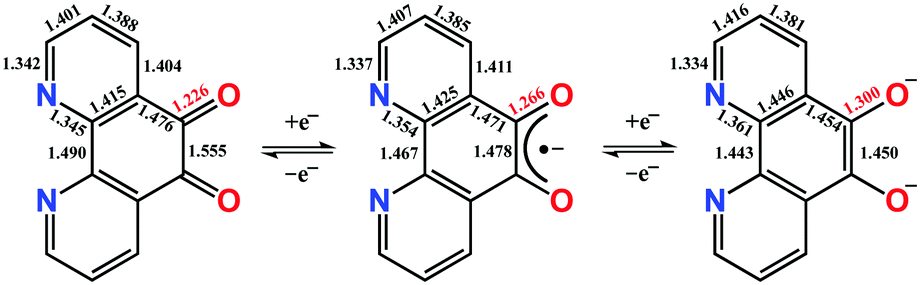 | ||
| Scheme 1 Three-membered electron transfer series for pd showing calculated bond distances. | ||
There is literature precedent for the formation of multi-metallic complexes using the dianionic form of the ligand.9–12 We also recently reported heterometallic rare earth-transition metal complexes utilising pd as a template.13 Our two-step route was necessary because although pd is reported to react with a number of reducing agents, the products are insoluble in common solvents, precluding collection of spectroscopic data and definitive assignment of redox state, but more importantly further reactivity.11,14,15
In contrast to the well-developed chemistry of transition metal semiquinones,16 there are only three reports of the in situ characterisation of pd˙−.17–19 These include two spectro-electrochemical studies in combination with EPR, and an EPR study.
Here we report the first organometallic synthons of the radical anion of pd, [CoCpR2]+[pd]˙− (R = H, (1); R = Me4, (2)). Furthermore, we demonstrate the coordination chemistry of 1 and 2, with [Y(hfac)3(thf)2] (hfac = 1,1,1-5,5,5-hexafluoroacetylacetonate) to synthesise the rare earth-transition metal heterometallic compounds, [CoCpR2]+[Y(hfac)3(N,N′-pd)]˙− (R = H, (3); R = Me4, (4)). Our first choice of synthon for this chemistry was the potassium salt of pd˙−, formed by reduction of pd with KH (see ESI†). However, while the spectroscopic data are consistent with [K]+[pd]˙−, its synthetic utility is limited. The reductants CoCpR2 were therefore selected to promote the formation of solvent separated ion pairs.
Complexes 1 and 2 were synthesised by the cold addition of a solution of CoCpR2 to a suspension of pd, both in acetonitrile. Complexes 1 and 2 were isolated after work-up as dark purple solids in good yields of 93% and 61%, respectively (Scheme 2). The lower yield of 2 is attributed to the CoCpt2 being generated in situ. The subsequent addition of a solution of [Y(hfac)3(thf)2] to a solution of either 1 (RT) or 2 (−35 °C) resulted in an immediate colour change from dark purple to dark green. After work-up, the heterometallic complexes 3 and 4 were isolated as dark green solids in good yields of 65% and 83%, respectively (Scheme 2). The purity of 1–4 were confirmed by elemental analysis and all complexes were fully characterised (see ESI† for full experimental details and spectra).
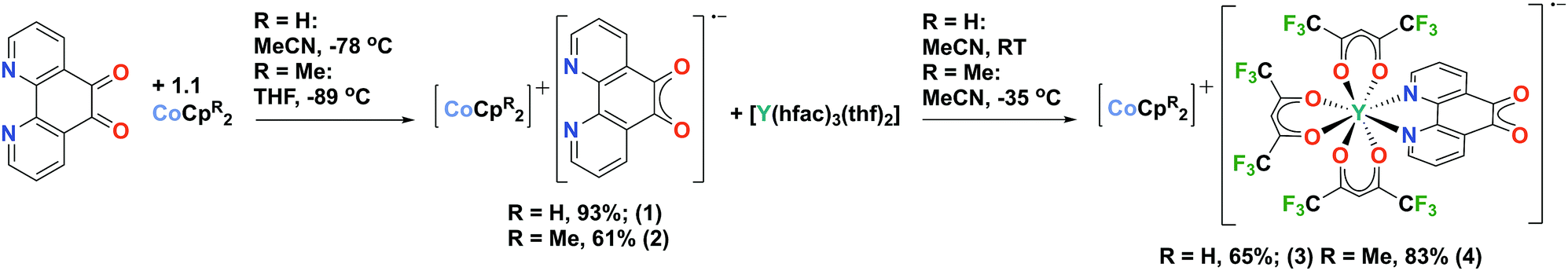 | ||
| Scheme 2 Synthetic route to radical anion containing rare-earth transition metal complexes. | ||
The 1H NMR spectra of 1 and 2 in d3-MeCN at room temperature are consistent with [CoCpR2]+ and display no other resonances. In addition to the [CoCpR2]+ resonances, the 1H NMR spectra of 3 in d3-MeCN and 4 in d8-THF at room temperature display both broadened proton environments in the pd˙− region and hfac singlets (this latter in the appropriate ratio for the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adduct). These data are consistent with remote i.e. N,N′-binding to pd˙−. The ATR IR spectra of 1 and 2 are also consistent with the reduction of pd to pd˙−. The νCO of neutral pd of 1678 cm−1 is no longer visible and there are two strong absorptions in the expected range for semiquinone radicals (1500–1400 cm−1) at 1511 cm−1 and 1485 cm−1.11,15 The ATR IR spectrum of [K(DME)x]+[pd]˙− (DME = dimethoxyethane, see ESI†), which does not contain aromatic νCC also displays two strong absorptions at 1518 cm−1 and 1504 cm−1. We therefore assign both absorptions in 1 and 2, centred ca. 1500 cm−1 to the symmetric and asymmetric stretch of the semiquinone. The ATR IR of 3 and 4 also contain multiple strong absorptions in the semiquinone region, however, their assignment cannot be definitive since the ancillary ligand vibrations from hfac also occur in the same region. There are no absorptions in catecholate region, consistent with N,N′-binding. Electronic spectra of 1–4 were collected in MeCN or THF (see ESI†) and are consistent with electrochemically generated pd˙−.17–19 Time dependent (TD) density functional (DFT) calculations have been used to assign the electronic spectra (vide infra).
:1 adduct). These data are consistent with remote i.e. N,N′-binding to pd˙−. The ATR IR spectra of 1 and 2 are also consistent with the reduction of pd to pd˙−. The νCO of neutral pd of 1678 cm−1 is no longer visible and there are two strong absorptions in the expected range for semiquinone radicals (1500–1400 cm−1) at 1511 cm−1 and 1485 cm−1.11,15 The ATR IR spectrum of [K(DME)x]+[pd]˙− (DME = dimethoxyethane, see ESI†), which does not contain aromatic νCC also displays two strong absorptions at 1518 cm−1 and 1504 cm−1. We therefore assign both absorptions in 1 and 2, centred ca. 1500 cm−1 to the symmetric and asymmetric stretch of the semiquinone. The ATR IR of 3 and 4 also contain multiple strong absorptions in the semiquinone region, however, their assignment cannot be definitive since the ancillary ligand vibrations from hfac also occur in the same region. There are no absorptions in catecholate region, consistent with N,N′-binding. Electronic spectra of 1–4 were collected in MeCN or THF (see ESI†) and are consistent with electrochemically generated pd˙−.17–19 Time dependent (TD) density functional (DFT) calculations have been used to assign the electronic spectra (vide infra).
Single crystals of 1 suitable for X-ray diffraction were grown from acetonitrile solution at −35 °C, over two weeks. The solid state molecular structure and selected parameters are shown in Fig. 1. These data represent the first structural data available for pd˙−. The metrics important for assessing the redox state of the ligand are those of the O,O′-pocket. In particular the bond distances of C5–O1 1.268(4) Å and C6–O2 1.278(4) Å in 1 are identical within error to one another and intermediate in length between a C–O double bond (pd0, C6–O2 1.23(2) and C5–O1 1.24(2) Å) and a C–O single bond (pd2−, C6–O2 1.35(1) and C5–O1 1.34(2) Å; Scheme 1). Likewise, the C5–C6 bond distance of 1.456(5) Å in 1, is shorter than the C–C single bond in pd0 (1.54(6) Å), but longer than the C–C double bond in pd2− (1.38(2) Å).13 These data are also in good agreement with the metrics obtained from DFT calculations (vide infra).
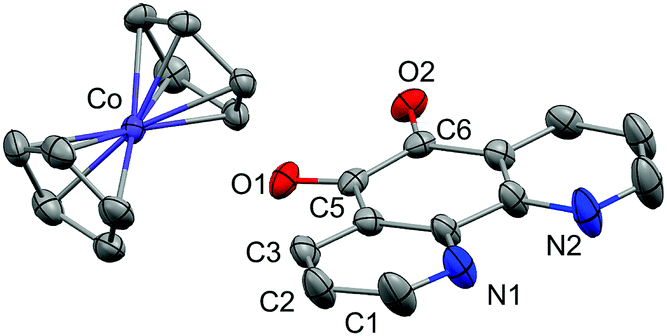 | ||
| Fig. 1 Molecular structure of 1. One ion pair of the four crystallographically independent pairs in the unit cell shown and hydrogen atoms omitted for clarity. Thermal ellipsoids drawn at 50% probability. Selected distances (Å): C5–C6 1.456(5), C5–O1 1.268(4), C6–O2 1.278(4), Co–C(Cp) 2.021(3)–2.034(4). | ||
We also obtained on one occasion the solid-state molecular structure of a protonated hydroquinone decomposition product [CoCp2]+[C12H7N2O2]− from the reaction of 1 with adventitious water. This decomposition product is crystallographically very different from 1 (see ESI†). We were unable to obtain single crystals of 3 and 4 suitable for X-ray diffraction. However, we assign the N,N′-binding mode in 3 and 4 on the basis of NMR, IR and EPR data. We see no evidence of linkage isomerism. Furthermore, the electronic spectra of 3 and 4 are well reproduced by the TD-DFT calculations (vide infra). We have therefore retained selectivity in binding the coordination chemistry precursor into the stronger donor N,N′-pocket, and thus directed future reactivity of this bridging ligand to the radical-O,O′-pocket.
To confirm the isolated radical form of pd, we measured the EPR spectra of 1 and 3 in MeCN solution at ambient temperature (Fig. 2). Spectral simulation of 1 was achieved with giso = 2.0053 consistent with a heterocyclic organic radical. The spectrum has rich hyperfine structure that was reproduced by including coupling to three pairs of protons in the ortho, meta and para positions of the aromatic ring (attached to C1–C3 in Fig. 1). These yielded couplings of 1.40, 1.08, and 1.56 × 10−4 cm−1, respectively. In addition, coupling to the 14N (I = 1) nuclei was estimated at 0.51 × 10−4 cm−1. These spin-Hamiltonian parameters are very similar to previously reported spectra of pd˙−.17–19 Coordination of the pd˙− to the YIII d0 ion results in a slightly wider spectrum with broader lines. The increase in line width is a consequence of slightly perturbed molecular tumbling commensurate with the inclusion of the bulky [Y(hfac)3] moiety. The centre of the spectra shifts to giso = 2.0041 with a minor change in the magnitude of the 1H hyperfine interaction to 1.41, 0.52, 1.56 × 10−4 cm−1 (cf.1 above), and an invariant 14N hyperfine splitting at 0.45 × 10−4 cm−1. Thus, the most profound change is the halving of the coupling to the proton in the meta position. The absence of 89Y (I = 1/2) coupling is likely a result of the polarised bonding with this Lewis acidic metal as well as its small nuclear moment (gN = −0.275) such that it is less than the experimental linewidth. Moreover, both spectra are consistent with a pd˙− radical where the spin is nearly exclusively localised to the O,O′ end of the molecule as supported by computational data.
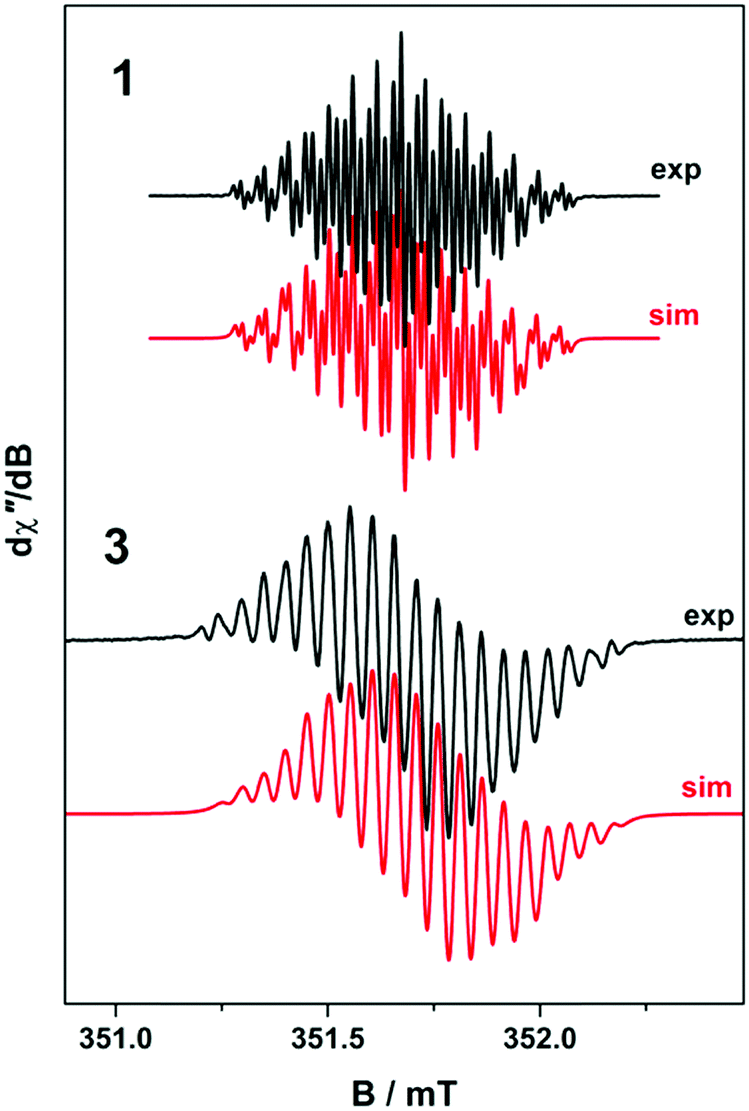 | ||
| Fig. 2 Comparison of the X-band EPR spectrum of 1 (experimental conditions: frequency, 9.8461 GHz; power, 0.63 mW; modulation, 0.007 mT), and 3 at 293 K (experimental conditions: frequency, 9.8653 GHz; power, 0.63 mW; modulation, 0.02 mT) recorded in MeCN solution at 293 K. Experimental data are represented by the black lines and simulation depicted by the red traces. | ||
The structure of the anion in 1 was geometry optimised using the BP86 functional and is consistent with the radical pd˙− (S = 1/2) form of the heterocycle (Scheme 1). The calculated C–O and C–C bond distances of the O,O′-pocket of 1.266 and 1.478 Å nicely match to the solid state structure (Fig. 1). In addition the C–C and C–N bond lengths exhibit a noticeable long-short-long pattern symptomatic of a quinoidal distortion commensurate with the addition of an unpaired electron to the aromatic system (Scheme 1).16,20 This effect diminishes the further the bond is from the O,O′-pocket. The Mulliken spin population analysis reveals 74% on the spin localised to this C2O2 unit, with a modest amount scattered over the phenanthroline part (Fig. S20, ESI†). The optimised structure of the complex ion in 3 depicts a square antiprism geometry adopted by the one pd˙− and three hfac− ligands coordinated to the central YIII ion (Fig. 3). The intraligand bond distances with the pd˙− are nearly identical to those observed for the free radical in 1. Its attachment to the YIII ion results in very little change in the structural change to the redox-active ligand. The Mulliken spin population shows an identical spin distribution for the coordinated pd˙− with the lion's share of the spin residing around the O,O′-pocket remote from the YIII centre (Fig. 3). No spin density is found on the YIII metal ion consistent with the weakly covalent bond diagnosed by EPR. These data are also consistent with N,N′-binding (vide supra) and in contrast to the perturbation to spin density and 89Y coupling, expected to result from O,O′-binding of the YIII centre.
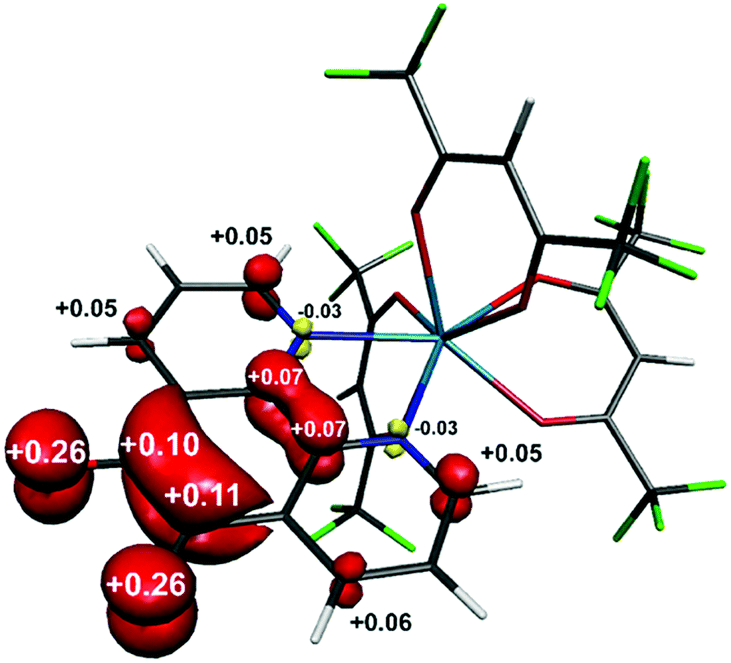 | ||
| Fig. 3 Mulliken spin density distribution in [Y(hfac)3(pd)]˙− (red: α-spin; yellow: β-spin). | ||
Intensely coloured solutions of 1 and 3 are the result of a dominant absorption band at 558 and 639 nm, respectively. To gain insight into the nature of this transition, we have calculated the electronic spectra based on the electronic structure for the anion in 1 and 3. Time-dependent (TD) DFT calculations were performed on the geometry optimised structures using MeCN as solvent in order to assign the electronic spectra. The vis-NIR spectra of both contain two transitions: 401 and 558 nm for 1, and 364 and 639 nm for 3 (Fig. 4). These bands are well reproduced, in particular the intensity, using the TD-DFT protocol though are blue-shifted ca. 1200 cm−1 compared to the experiment. For 1, the bands which are π → π* transitions appear at 411 and 522 nm, result from HOMO−2 → SOMO and SOMO → LUMO+1 excitations, respectively (Fig. S22, ESI†); the latter MOs are shown in the inset in Fig. 4. The analogous bands in 3 are calculated at 405 and 567 nm, respectively (Fig. 4). These are also assigned as π → π* transitions within the pd˙−, although in this instance as the organic radical is coordinated to a metal ion, they are specified as intraligand charge transfer (ILCT) transitions. Analysis of the donor and acceptor MOs shows the band at 567 is the SOMO → LUMO+4 excitation, shown in the inset of Fig. 4. The higher energy band at 405 nm is predominantly a HOMO−2 → LUMO transition (Fig. S23, ESI†); the order is different to 1 as it includes contributions from the [Y(hfac)3] moiety. Again, the intensity of these two transitions is nicely reproduced by the TD-DFT calculations. These transitions are diagnostic of the singly-reduced radical pd˙−,17–19 as also observed for related 9,10-phenanthrenesemiquinone (pq˙−) complexes [MX(pq˙−)(PPh3)2(CO)] (M = Ru, X = Cl; M = Os, X = Br) where the dominant feature is also assigned as a π → π* ILCT.21,22
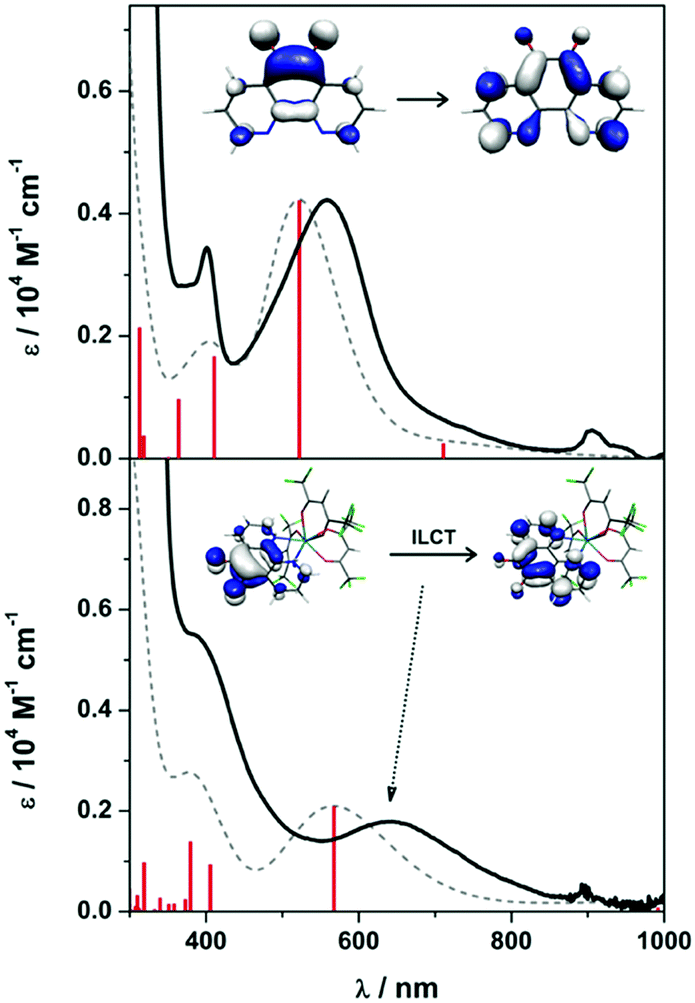 | ||
| Fig. 4 Overlay of the experimental (solid line) and calculated (dashed line) electronic absorption spectra for 1 (top) and 3 (bottom) recorded at ambient temperature in MeCN. Vertical bars represent individual calculated transitions. Inset orbitals depict the donor/acceptor molecular orbitals (MO) that constitute the band at 558 nm in 1 and 639 nm in 3. | ||
In conclusion, this work represents the discovery and success of a novel approach to the synthesis of rare earth-transition metal complexes. The stability of the bridging pd˙− ligand, and the selectivity of N,N′-binding for Y(III) have been demonstrated. It is anticipated that compounds 1–4 will be useful synthons in pursuit of mixed-metal radical-bridged f-element complexes in the future.
The authors acknowledge the EPSRC (EP/L504786/1 and EP/M508056/1) and the University of Glasgow for funding. We also acknowledge the EPSRC UK National Crystallography Service at the University of Southampton.
Conflicts of interest
There are no conflicts to declare.Notes and references
- D. Astruc, Electron Transfer and Radical Processes in Transition-Metal Chemistry, Wiley VCH, 1995 Search PubMed.
- P. J. Chirik and K. Wieghardt, Science, 2010, 327, 794–795 CrossRef PubMed.
- N. H. Anderson, S. O. Odoh, Y. Yao, U. J. Williams, B. A. Schaefer, J. J. Kiernicki, A. J. Lewis, M. D. Goshert, P. E. Fanwick, E. J. Schelter, J. R. Walensky, L. Gagliardi and S. C. Bart, Nat. Chem., 2014, 6, 919 CrossRef PubMed.
- M. Murugesu and E. J. Schelter, Inorg. Chem., 2016, 55, 9951–9953 CrossRef PubMed.
- P. L. Arnold and Z. R. Turner, Nat. Rev. Chem., 2017, 1, 0002 CrossRef.
- H. S. L. Pierre and K. Meyer, in Prog. Inorg. Chem., ed. K. D. Karlin, 2014, vol. 58, pp. 303–416 Search PubMed.
- W. J. Evans, M. Fang, J. E. Bates, F. Furche, J. W. Ziller, M. D. Kiesz and J. I. Zink, Nat. Chem., 2010, 2, 644 CrossRef PubMed.
- S. Demir, L.-R. Jeon, J. R. Long and T. D. Harris, Coord. Chem. Rev., 2015, 289–290, 149–176 CrossRef.
- G. A. Fox, S. Bhattacharya and C. G. Pierpont, Inorg. Chem., 1991, 30, 2895–2899 CrossRef.
- A. Y. Girgis, Y. S. Sohn and A. L. Balch, Inorg. Chem., 1975, 14, 2327–2331 CrossRef.
- F. Calderazzo, F. Marchetti, G. Pampaloni and V. Passarelli, J. Chem. Soc., Dalton Trans., 1999, 4389–4396 RSC.
- N. M. Shavaleev, L. P. Moorcraft, S. J. A. Pope, Z. R. Bell, S. Faulkner and M. D. Ward, Chem. Commun., 2003, 1134–1135 RSC.
- J. R. Hickson, S. J. Horsewill, C. Bamforth, J. McGuire, C. Wilson, S. Sproules and J. H. Farnaby, Dalton Trans., 2018, 47, 10692–10701 RSC.
- E. K. Brechin, L. Calucci, U. Englert, L. Margheriti, G. Pampaloni, C. Pinzino and A. Prescimone, Inorg. Chim. Acta, 2008, 361, 2375–2384 CrossRef.
- F. Calderazzo, G. Pampaloni and V. Passarelli, Inorg. Chim. Acta, 2002, 330, 136–142 CrossRef.
- C. G. Pierpont and C. W. Lange, in Prog. Inorg. Chem., 2007, pp. 331–442 Search PubMed.
- D. M. Murphy, K. McNamara, P. Richardson, V. Sanchez-Romaguera, R. E. P. Winpenny and L. J. Yellowlees, Inorg. Chim. Acta, 2011, 374, 435–441 CrossRef.
- H. Bock and P. Hänel, Z. Naturforsch. B, 1992, 47, 288 Search PubMed.
- S. Berger, J. Fiedler, R. Reinhardt and W. Kaim, Inorg. Chem., 2004, 43, 1530–1538 CrossRef PubMed.
- S. Sproules and K. Wieghardt, Coord. Chem. Rev., 2011, 255, 837–860 CrossRef.
- M. K. Biswas, S. C. Patra, A. N. Maity, S.-C. Ke, N. D. Adhikary and P. Ghosh, Inorg. Chem., 2012, 51, 6687–6699 CrossRef PubMed.
- M. K. Biswas, S. C. Patra, A. N. Maity, S.-C. Ke, T. Weyhermuller and P. Ghosh, Dalton Trans., 2013, 42, 6538–6552 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1854232 and 1854233. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc05591b |
| This journal is © The Royal Society of Chemistry 2018 |