
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZ-Schematic and visible-light-driven CO2 reduction using H2O as an electron donor by a particulate mixture of a Ru-complex/(CuGa)1−xZn2xS2 hybrid catalyst, BiVO4 and an electron mediator†
Tomiko M.
Suzuki
 *a,
Shunya
Yoshino
b,
Tomoaki
Takayama
b,
Akihide
Iwase
b,
Akihiko
Kudo
*b and
Takeshi
Morikawa
a
*a,
Shunya
Yoshino
b,
Tomoaki
Takayama
b,
Akihide
Iwase
b,
Akihiko
Kudo
*b and
Takeshi
Morikawa
a
aToyota Central R&D Labs., Inc., 41-1 Yokomichi, Nagakute, Aichi 480-1192, Japan. E-mail: tomiko@mosk.tytlabs.co.jp
bDepartment of Applied Chemistry, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: a-kudo@rs.kagu.tus.ac.jp
First published on 14th August 2018
Abstract
Visible-light-driven Z-schematic CO2 reduction using H2O as an electron donor was achieved using a simple mixture of a metal-sulfide/molecular hybrid photocatalyst for CO2 reduction, a water oxidation photocatalyst and a redox-shuttle electron mediator. This is the first demonstration of a highly selective particulate CO2 reduction system accompanying O2 generation utilizing a semiconductor/molecular hybrid photocatalyst.
Photocatalytic CO2 reduction into useful energy-rich chemicals using water as an electron donor with a particulate system has attracted attention for sustainable and scalable artificial photosynthesis to mitigate global warming and generate useful fuels.1–5 Particulate systems are recognized as cost-effective and could be the ultimate tool for CO2 fixation and solar fuel generation.6,7 A photocatalytic Z-scheme (or two-step photoexcitation) system that connects photocatalysts for CO2 reduction with H2O oxidation is considered to be a promising approach; however, few particulate Z-scheme systems for CO2 reduction using water as an electron donor have been reported. Visible-light-driven conversion of CO2 into CO over a combined system of CoOx-loaded BiVO4 and metal sulfides with a reduced graphene oxide electron mediator has yet to reach 1% CO selectivity. This is due to competitive H2 generation and low CO2 selectivity at metal sulfide surfaces.8,9
A hybrid catalyst of a semiconductor10–12 linked with a metal-complex catalyst13–15 is promising for visible-light-driven selective CO2 reduction with regard to high selectivity, depending on the selective coordination of CO2 molecules to the metal centers of the complexes.13,16–22 It is essential that photoexcited electrons in the conduction band (CB) of the semiconductor transfer to the metal-complex catalyst within the picosecond region, which leads to two-electron reduction of CO2 at the complex.23 We previously demonstrated solar formate generation from CO2 and H2O using a photocathode of a Ru-complex catalyst linked with Zn-doped InP or N,Zn-codoped Fe2O3 combined with TiO2 or SrTiO3−x photoanodes,24–26 and confirmed the potential of the system with a solar-to-chemical conversion efficiency of 4.6% over a monolithic Ru-complex-polymer/Si–Ge/IrOx electrode.27 Ishitani and colleagues reported photoelectrochemical CO2 reduction to CO via H2O oxidation using a photocathode composed of a Ru(II)–Re(I) supramolecular metal complex immobilized on a NiO or CuGaO2 semiconductor.28,29 In contrast, a particulate system for Z-schematic CO2 reduction using a metal complex combined with a H2O oxidation reaction has not yet been demonstrated. Efficient electron transfer from the H2O oxidation site to the CO2 reduction site without degradation of the catalytic activity for both reactions should be implemented.
To realize Z-schematic CO2 reduction with a particulate system, we focused our attention on metal sulfides as the CO2 reduction semiconductor because they generally possess long photoexcited carrier lifetimes and narrow band gaps (BGs) originating from the S 3p state, which allows absorption of a substantial amount of visible light. Reisner et al. reported photocatalytic conversion of CO2 into CO in water with >90% selectivity over a Ni–cyclam catalyst anchored with CdS.19 We reported CO2 photoconversion into HCOOH over ZnS:Ni and (AgIn)0.22Zn1.56S2 linked with a Ru–bipyridine catalyst [Ru(4,4′-diphosphonate-2,2′-bipyridine)(CO)2Cl2] ([Ru(dpbpy)]).30 These Cd- and Zn-based sulfides are considered to be beneficial when combined with molecular catalysts because of their negative CB minimum (ECBM) positions originating from Cd 5s5p and Zn 4s4p orbitals, respectively, which facilitate fast electron transfer from the CB in a photoexcited state to a metal-complex catalyst due to the greater energy difference (ΔG) between the ECBM and CO2 reduction potential of the metal-complex, which promotes the CO2 reduction reaction. However, these systems conducted half reactions that required triethanolamine as a strong sacrificial electron donor. Here, we demonstrate the first particulate-based visible-light-driven Z-scheme system that harmonizes both reactions of CO2 reduction and H2O oxidation over a hybrid photocatalyst of a semiconductor/molecular-catalyst.
Fig. 1(a) shows an illustration of the Z-schematic system for photoconversion of CO2 to CO and HCOO− over [Ru(dpbpy)]-modified (CuGa)1−xZn2xS2 as a CO2 reduction hybrid photocatalyst, [Co(tpy)2]3+/2+ (tpy: 2,2′:6,2′′-terpyridine) as an electron mediator, and BiVO4 as a water oxidation photocatalyst. Fig. 1(b) shows that the simple mixture in an aqueous solution produced CO and HCOO− at almost linear rates together with O2 generation under visible light irradiation after hours of pre-irradiation in a CO2 flow reactor,8,9i.e., the Z-scheme reaction. Although BiVO4 can photogenerate O2, ECBM for BiVO4 is located at an unfavorably deep position for the reduction reaction of CO2 and protons.8,31 Thus, the CO2 reduction reaction could occur at the [Ru(dpbpy)]/(CuGa)0.3Zn1.4S2 photocatalyst, which suggests a Z-scheme mechanism with H2O as an electron donor, which is discussed later. Although H2 evolution by proton reduction also proceeded, the CO2 reduction selectivity to total reductive products (CO, HCOO−, H2) for 9 h reached 64 mol%, significantly higher than the ca. 1% of previous non-molecular particulate Z-scheme systems.8,9 The modification of [Ru(dpbpy)] led to the production of HCOO− and the turnover number (TON) of HCOO− evolution was calculated to be 17, assuming that all [Ru(dpbpy)] remained on (CuGa)1−xZn2xS2 during the photocatalytic reaction.
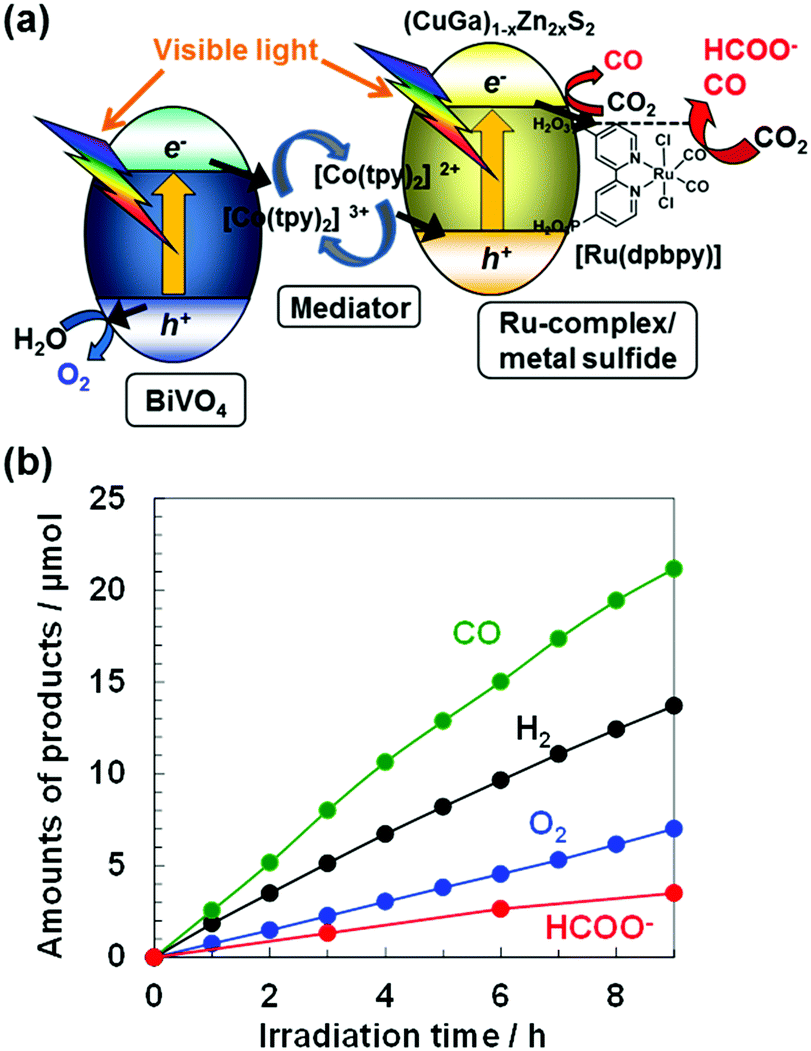 | ||
| Fig. 1 (a) Visible-light-driven Z-schematic system for CO2 reduction consisting of particulate [Ru(dpbpy)] modified (CuGa)1−xZn2xS2 hybrid photocatalysts, a BiVO4 photocatalyst, and a [Co(tpy)2]3+/2+ redox shuttle electron mediator. (b) Z-schematic CO2 reduction in an aqueous solution with a CO2 flow system. Conditions: [Ru(dpbpy)]/(CuGa)0.3Zn1.4S2 (0.4 g) and BiVO4 (0.2 g); 0.02 mM [Co(tpy)2]2+ containing 0.1 M NaHCO3 aqueous solution (150 mL) and visible-light (λ > 420 nm). | ||
The key to designing the visible-light-driven Z-schematic system is the hybrid photocatalyst for CO2 reduction. Thus, a sufficient energy difference ΔG to facilitate fast electron transfer from the semiconductor to the Ru-complex catalyst plays a decisive role.16,23 Electron transfer from the water oxidation photocatalyst to the CO2 reduction photocatalyst is also indispensable. First, the optimal combination of Ru-complex and semiconductor was investigated. A solid solution of CuGaS2 and ZnS ((CuGa)1−xZn2xS2)32 was selected as the semiconductor for the hybrid photocatalyst because their BG and band position, particularly ECBM, are dominated by Zn 4s4p + Ga 4s4p and are tunable by changing x. This could be beneficial to control the electron transfer rate from the CB to the molecular catalysts. XRD measurements of (CuGa)1−xZn2xS2 synthesized by a solid-state reaction32 revealed that a single chalcopyrite phase (x ≤ 0.2) or zincblende structure (x ≥ 0.5) was obtained (Fig. S1 and Table S1, ESI†). The BG of CuGaS2 and (CuGa)1−xZn2xS2 (x < 1.0) was within the range of 2.24–2.54 eV, which enables absorption of visible light (Fig. S2 and Table S1, ESI†). Hybrid catalysts of 0.03–0.08 wt% [Ru(dpbpy)] with the phosphonate ligand (CO2 reduction potential of ca. −1.0 V vs. NHE)30 linked with (CuGa)1−xZn2xS2 were prepared using an adsorption method30 (Table S2, ESI†).
Typical Z-schematic CO2 reduction was performed by using [Ru(dpbpy)]/(CuGa)1−xZn2xS2 and BiVO4 in a test tube filled with [Co(tpy)2]2+ containing aqueous NaHCO3 solution saturated with CO2 under visible light irradiation for 16 h. This method is useful to measure the tendency of the sample-dependent product for multiple runs.33Fig. 2(a) shows that the CO2 reduction activity was significantly dependent on the composition of (CuGa)1−xZn2xS2. For bare (CuGa)1−xZn2xS2, the amount of CO produced increased with x within the range of 0.0 ≤ x ≤ 0.5, which could be explained by the more negative ECBM and narrower BG (Fig. 2(b) and Fig. S2, ESI†), which led to a more efficient electron transfer toward a higher CO2 reduction rate. Further increase in x (x ≥ 0.7) decreased CO evolution due to a reduced amount of absorbed photons with a wider BG (≥2.36 eV). H2 evolution induced by the competitive proton reduction increased with x within the range of 0.5 ≤ x ≤ 0.7. Replacement of the bare (CuGa)1−xZn2xS2 with the [Ru(dpbpy)]/(CuGa)1−xZn2xS2 hybrid photocatalyst not only enhanced the CO formation rate but also induced formate production; [Ru(dpbpy)] is known as a catalyst for the production of formate and CO.30,34 [Ru(dpbpy)]/(CuGa)1−xZn2xS2 at x = 0.7 showed the highest CO2 reduction activity for CO and HCOO− production, with TONs (based on product generated by the [Ru(dpbpy)] catalyst) of 214 and 70 after 16 h of reaction, respectively. In the Z-schematic water splitting system composed of metallic Ru-loaded (CuGa)1−xZn2xS2 for H2 generation, CoOx-loaded BiVO4 for water oxidation and [Co(tpy)2]2+/3+ for electron mediation, the highest water splitting rate was reported at x = 0.2.32 In contrast, [Ru(dpbpy)]/(CuGa)0.8Zn0.4S2 (x = 0.2) showed a negligibly small CO2 reduction activity. These results suggest that more negative ECBM (with greater x) improves the electron transfer rate for CO2 reduction at [Ru(dpbpy)].16,17,23,30 The CO2 reduction activity of the hybrid catalyst is in a trade-off relationship between the negative ECBM to facilitate electron transfer to the complex catalyst and the BG to determine the number of photons absorbed; therefore, overall matching in the electron transfer process was successful in the present Z-schematic ([Ru(dpbpy)]/(CuGa)1−xZn2xS2)–([Co(tpy)2]3+/2+)–(BiVO4) system.
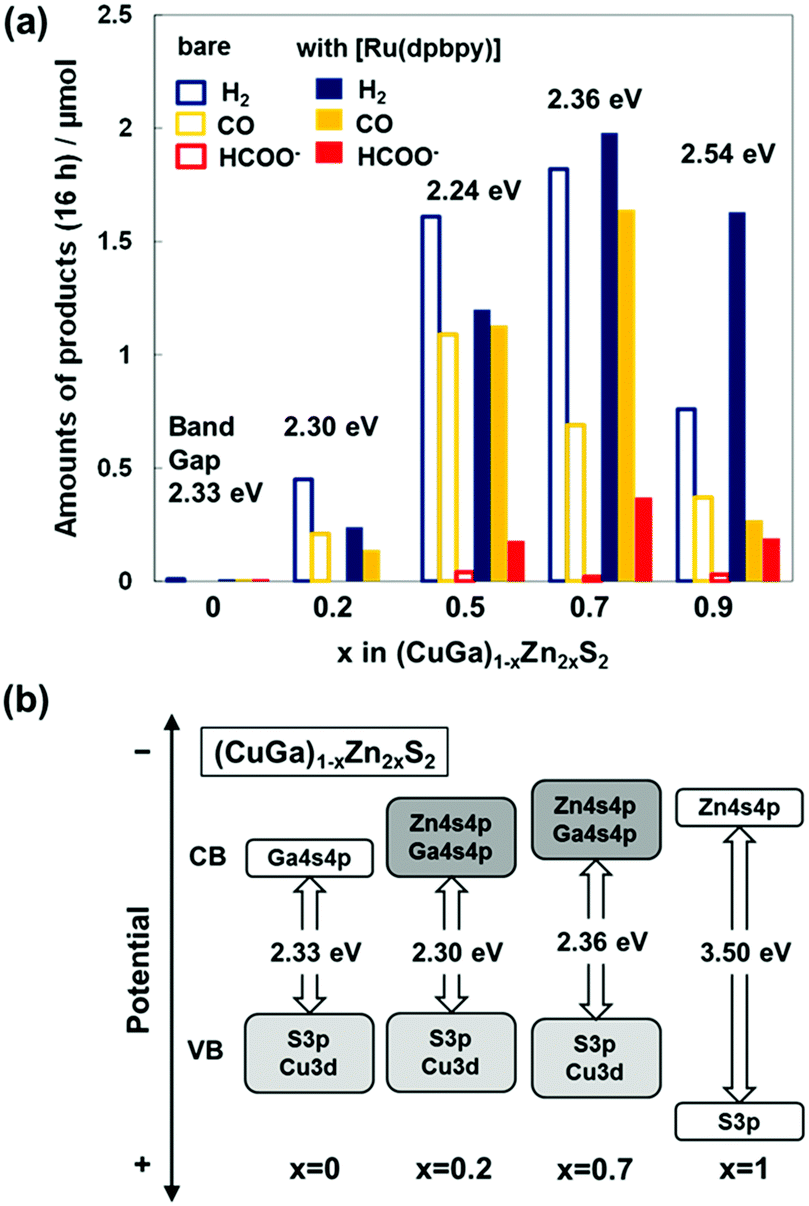 | ||
| Fig. 2 (a) Z-Schematic CO2 reduction using the (CuGa)1−xZn2xS2 or ([Ru(dpbpy)]/(CuGa)1−xZn2xS2)–([Co(tpy)2]3+/2+)–(BiVO4) systems under visible-light irradiation using the test tube method. Conditions: 8 mg of each photocatalyst; 0.02 mM [Co(tpy)2]2+ containing 0.1 M NaHCO3 aqueous solution (4 mL); visible-light (390 < λ ≤ 750 nm) for 16 h. (b) Estimated band structures of (CuGa)1−xZn2xS2. | ||
Experimental data used to determine the effective components for the Z-schematic CO2 reduction are summarized in Table 1. A mixture of BiVO4 and [Co(tpy)2]2+ produced neither H2, nor CO, nor HCOO− due to the unfavorably deep ECBM position (entry 1). The addition of (CuGa)0.3Zn1.4S2 produced H2 and CO (entry 2), and [Ru(dpbpy)] modification of (CuGa)0.3Zn1.4S2 induced HCOO− formation and further improved CO production by the effect of [Ru(dpbpy)] (entry 3, Fig. S3, ESI†). The absence of [Co(tpy)2]2+ largely decreased CO and HCOO− generation (entry 4), and removal of BiVO4 resulted in the termination of both CO and HCOO− formation (entry 5, Fig. S4, ESI†). These results indicate that BiVO4 is necessary for high CO2 reduction activity, and the presence of [Co(tpy)2]2+ is essential for a high CO and HCOO− formation rate. The absence of NaHCO3 decreased the formation of both CO and HCOO− (entry 6), and the formation of CO and HCOO− was negligible in the absence of [Co(tpy)2]2+ (entry 7). Among the aqueous electrolyte and Co-complexes, NaHCO3 and [Co(tpy)2]2+ were evaluated to be the best for the CO2 reduction selectivity and production rate, respectively (Tables S3 and S4, ESI†). It was also confirmed that no reaction occurred without irradiation (entry 8), and that the formation of CO and HCOO− was negligibly small when Ar was bubbled in the solution (entry 9).
| Entry | CO2 reduction photocatalyst | Mediator | O2 evolution photocatalyst | Salt | Gas | hν | Amount of products (μmol) | |||
|---|---|---|---|---|---|---|---|---|---|---|
| (CuGa)0.3Zn1.4S2 | [Ru(dpbpy)] | [Co(tpy)2]2+ | BiVO4 | NaHCO3 | H2 | CO | HCOO− | |||
| a Conditions: 8 mg of each photocatalyst; 0.02 mM [Co(tpy)2]2+ containing 0.1 M NaHCO3 aqueous solution (4 mL); visible-light (390 < λ ≤ 750 nm) for 16 h; Pyrex test tube. | ||||||||||
| 1 | ✓ | ✓ | ✓ | CO2 | ✓ | 0.00 | 0.00 | 0.00 | ||
| 2 | ✓ | ✓ | ✓ | ✓ | CO2 | ✓ | 1.82 | 0.69 | 0.02 | |
| 3 | ✓ | ✓ | ✓ | ✓ | ✓ | CO2 | ✓ | 1.98 | 1.64 | 0.37 |
| 4 | ✓ | ✓ | ✓ | ✓ | CO2 | ✓ | 1.53 | 0.10 | 0.10 | |
| 5 | ✓ | ✓ | ✓ | ✓ | CO2 | ✓ | 1.06 | 0.79 | 0.28 | |
| 6 | ✓ | ✓ | ✓ | ✓ | CO2 | ✓ | 3.59 | 0.56 | 0.18 | |
| 7 | ✓ | ✓ | ✓ | CO2 | ✓ | 2.42 | 0.01 | 0.02 | ||
| 8 | ✓ | ✓ | ✓ | ✓ | ✓ | CO2 | 0.00 | 0.00 | 0.00 | |
| 9 | ✓ | ✓ | ✓ | ✓ | ✓ | Ar | ✓ | 0.96 | 0.01 | 0.01 |
[Co(tpy)2]2+ was reported to act as a [Co(tpy)2]3+/2+ redox shuttle electron mediator connecting two semiconductors for H2 and O2 generation in Z-scheme water splitting.32 As inferred from the mechanism of the IO3−/I− shuttle redox mediator system,35,36 it is speculated that [Co(tpy)2]3+ may favorably adsorb onto BiVO4, while [Co(tpy)2]2+ adsorbs onto (CuGa)0.3Zn1.4S2, mediating electrons between them as a redox shuttle. [Co(tpy)2]2+ was reported to be an electrocatalyst for the reduction of CO2 to CO in dimethylformamide/H2O (90/10 v/v) when one terpyridine ligand was eliminated.37,38 However, the intrinsic potential for the reaction is too negative (−2.0 V vs. Fc/Fc+) compared to the ECBM of (CuGa)0.3Zn1.4S2; therefore, [Co(tpy)2]2+ is not a catalyst in the present case. [Co(tpy)2]2+ did not catalyze CO2 reduction in an aqueous solution when electrically biased in the ECBM region under visible light irradiation.
Isotope tracer analyses using 13CO2 confirmed that the carbon source of evolved CO and HCOO− was dissolved CO2 (Fig. S5 and S6, ESI†). O2 was also confirmed to originate from water using H218O (Fig. S7, ESI†), in which the total amount of 18O2 and 16O18O was more than 85% of the total dioxygen detected. These results explained that CO2 was reduced to CO and HCOO− using electrons extracted from H2O molecules. The TON for O2 evolution with the Co-complex was calculated to be 9 (9 h), which suggests that the Co-complex acted as an electron mediator. A slight deviation from stoichiometric O2 evolution (4-electron reaction) compared with half of the total of CO + HCOO− + H2 (2-electron reactions) in Fig. 1(b) was similarly observed in all-inorganic Z-scheme systems for CO2 reduction.8,9 Self-photooxidation of (CuGa)1−xZn2xS2 could partially supply electrons for the CO2 reduction reaction. Further investigations will clarify the overall electron/hole stoichiometry. X-ray photoelectron spectroscopy (XPS) measurements before and after the Z-scheme reaction (in Fig. 1(b)) revealed no change in the chemical state of Ru and sulfur ions (Fig. S8, ESI†). Half of the amount of [Ru(dpbpy)] was eliminated from the (CuGa)1−xZn2xS2 surface according to the change in the Ru/S and Ru/Zn ratios (Table S5, ESI†).
In conclusion, a visible-light-driven Z-schematic CO2 reduction to CO and HCOO− in an aqueous particulate suspension system was achieved using a simple mixture of [Ru(dpbpy)]/(CuGa)1−xZn2xS2 hybrid, [Co(tpy)2]2+ and BiVO4. Adjustment of band alignment is essential to the Z-schematic CO2 reduction reaction accompanying O2 generation. The very high CO2 reduction selectivity beyond 60% against competing H2 generation strongly suggests that the particulate Z-schematic system is feasible to construct selective and efficient photocatalysts for CO2 fixation and solar fuel generation.
This work was partially supported by the JST ACT-C Grant Number JPMJCR12ZA, Japan (T. M. S. and T. M.). The authors thank Ms Ayako Oshima, Mr Kosuke Kitazumi, Ms Naoko Takahashi, Mr Satoru Kosaka, Mr Ikoma Narita and Ms Saori Narita for experimental assistance. The authors also thank Dr Shunsuke Sato, Dr Keita Sekizawa, and Dr Hiromitsu Tanaka for fruitful discussions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- W. Kim, G. Yuan, B. A. McClure and H. Frei, J. Am. Chem. Soc., 2014, 136, 11034–11042 CrossRef PubMed.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. Kenis, C. A. Kerfeld, R. H. Morris, C. H. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef PubMed.
- J. L. White, M. F. Baruch, J. E. Pander III, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef PubMed.
- H. Dau, E. Fujita and L. Sun, ChemSusChem, 2017, 10, 4228–4235 CrossRef PubMed.
- M. Wang, V. Artero, L. Hammarstrom, J. Martinez, J. Karlsson, D. Gust, P. Summers, C. Machan, P. Brueggeller, C. D. Windle, Y. Kageshima, R. Cogdell, K. R. Tolod, A. Kibler, D. H. Apaydin, E. Fujita, J. Ehrmaier, S. Shima, E. Gibson, F. Karadas, A. Harriman, H. Inoue, A. Kudo, T. Takayama, M. Wasielewski, F. Cassiola, M. Yagi, H. Ishida, F. Franco, S. O. Kang, D. Nocera, C. Li, F. Di Fonzo, H. Park, L. Sun, T. Setoyama, Y. S. Kang, O. Ishitani, J. R. Shen, H. J. Son and S. Masaoka, Faraday Discuss., 2017, 198, 353–395 RSC.
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983–2002 RSC.
- D. M. Fabian, S. Hu, N. Singh, F. A. Houle, T. Hisatomi, K. Domen, F. E. Osterloh and S. Ardo, Energy Environ. Sci., 2015, 8, 2825–2850 RSC.
- A. Iwase, S. Yoshino, T. Takayama, Y. H. Ng, R. Amal and A. Kudo, J. Am. Chem. Soc., 2016, 138, 10260–10264 CrossRef PubMed.
- T. Takayama, K. Sato, T. Fujimura, Y. Kojima, A. Iwase and A. Kudo, Faraday Discuss., 2017, 198, 397–407 RSC.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- W. Kim, T. Seok and W. Choi, Energy Environ. Sci., 2012, 5, 6066–6070 RSC.
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef.
- G. Sahara and O. Ishitani, Inorg. Chem., 2015, 54, 5096–5104 CrossRef PubMed.
- C. Costentin, M. Robert, J. M. Saveant and A. Tatin, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6882–6886 CrossRef PubMed.
- H. Takeda, C. Cometto, O. Ishitani and M. Robert, ACS Catal., 2017, 7, 70–88 CrossRef.
- S. Sato, T. Morikawa, S. Saeki, T. Kajino and T. Motohiro, Angew. Chem., Int. Ed., 2010, 49, 5101–5105 CrossRef PubMed.
- T. M. Suzuki, H. Tanaka, T. Morikawa, M. Iwaki, S. Sato, S. Saeki, M. Inoue, T. Kajino and T. Motohiro, Chem. Commun., 2011, 47, 8673–8675 RSC.
- R. Kuriki, M. Yamamoto, K. Higuchi, Y. Yamamoto, M. Akatsuka, D. Lu, S. Yagi, T. Yoshida, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2017, 56, 4867–4871 CrossRef PubMed.
- M. F. Kuehnel, K. L. Orchard, K. E. Dalle and E. Reisner, J. Am. Chem. Soc., 2017, 139, 7217–7223 CrossRef PubMed.
- M. F. Kuehnel, C. D. Sahm, G. Neri, J. R. Lee, K. L. Orchard, A. J. Cowan and E. Reisner, Chem. Sci., 2018, 9, 2501–2509 RSC.
- H. N. Tian, ChemSusChem, 2015, 8, 3746–3759 CrossRef PubMed.
- K. Sekizawa, K. Maeda, K. Domen, K. Koike and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 4596–4599 CrossRef PubMed.
- K. Yamanaka, S. Sato, M. Iwaki, T. Kajino and T. Morikawa, J. Phys. Chem. C, 2011, 115, 18348–18353 CrossRef.
- S. Sato, T. Arai, T. Morikawa, K. Uemura, T. M. Suzuki, H. Tanaka and T. Kajino, J. Am. Chem. Soc., 2011, 133, 15240–15243 CrossRef PubMed.
- T. Arai, S. Sato, T. Kajino and T. Morikawa, Energy Environ. Sci., 2013, 6 Search PubMed.
- K. Sekizawa, S. Sato, T. Arai and T. Morikawa, ACS Catal., 2018, 8, 1405–1416 CrossRef.
- T. Arai, S. Sato and T. Morikawa, Energy Environ. Sci., 2015, 8, 1998–2002 RSC.
- G. Sahara, H. Kumagai, K. Maeda, N. Kaeffer, V. Artero, M. Higashi, R. Abe and O. Ishitani, J. Am. Chem. Soc., 2016, 138, 14152–14158 CrossRef PubMed.
- H. Kumagai, G. Sahara, K. Maeda, M. Higashi, R. Abe and O. Ishitani, Chem. Sci., 2017, 8, 4242–4249 RSC.
- T. M. Suzuki, T. Takayama, S. Sato, A. Iwase, A. Kudo and T. Morikawa, Appl. Catal., B, 2018, 224, 572–578 CrossRef.
- Y. Xu, S. Wang, J. Yang, B. Han, R. Nie, J. Wang, J. Wang and H. Jing, Nano Energy, 2018, 51, 442–450 CrossRef.
- T. Kato, Y. Hakari, S. Ikeda, Q. Jia, A. Iwase and A. Kudo, J. Phys. Chem. Lett., 2015, 6, 1042–1047 CrossRef PubMed.
- T. M. Suzuki, A. Iwase, H. Tanaka, S. Sato, A. Kudo and T. Morikawa, J. Mater. Chem. A, 2015, 3, 13283–13290 RSC.
- Y. Kuramochi, J. Itabashi, K. Fukaya, A. Enomoto, M. Yoshida and H. Ishida, Chem. Sci., 2015, 6, 3063–3074 RSC.
- R. Abe, K. Sayama, K. Domen and H. Arakawa, Chem. Phys. Lett., 2001, 344, 339–344 CrossRef.
- R. Abe, Bull. Chem. Soc. Jpn., 2011, 84, 1000–1030 CrossRef.
- N. Elgrishi, M. B. Chambers, V. Artero and M. Fontecave, Phys. Chem. Chem. Phys., 2014, 16, 13635–13644 RSC.
- N. Elgrishi, M. B. Chambers and M. Fontecave, Chem. Sci., 2015, 6, 2522–2531 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, XRD patterns, UV-vis diffuse reflectance spectra, Z-schematic CO2 reduction using a hybrid photocatalyst, BiVO4 and a Co-complex, GC-MS chromatograms, IC-TOFMS chromatograms, and XPS spectra. See DOI: 10.1039/c8cc05505j |
| This journal is © The Royal Society of Chemistry 2018 |