
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thiazolidine chemistry revisited: a fast, efficient and stable click-type reaction at physiological pH†
Daniel
Bermejo-Velasco
a,
Ganesh N.
Nawale
 a,
Oommen P.
Oommen
b,
Jöns
Hilborn
a and
Oommen P.
Varghese
*a
a,
Oommen P.
Oommen
b,
Jöns
Hilborn
a and
Oommen P.
Varghese
*a
aTranslational Chemical Biology Laboratory, Division of Polymer Chemistry, Department of Chemistry-Ångstrom, Uppsala University, Uppsala, Sweden. E-mail: oommen.varghese@kemi.uu.se
bBioengineering and Nanomedicine Lab, Faculty of Biomedical Sciences and Engineering, Tampere University of Technology, and BioMediTech Institute, 33720, Tampere, Finland
First published on 16th October 2018
Abstract
We describe the fast reaction kinetics between 1,2-aminothiols and aldehydes. Under physiological conditions such a click-type reaction afforded a thiazolidine product that remains stable and did not require any catalyst. This type of bioorthogonal reaction offers enormous potential for the coupling of biomolecules in an efficient and biocompatible manner.
Conjugation strategies for biomolecules require the design of highly chemoselective, specific and fast bioorthogonal reactions that can produce stable conjugates under physiological conditions without employing any toxic reagents or producing any by-products.1–3 In recent years, bioconjugation of functional molecules has received significant attention due to the emerging field of biomedicine that requires covalent conjugation of biomolecules such as nucleic acids, antibodies, proteins as well as glycosaminoglycans to study and manipulate cellular processes.4–7 For this purpose, the most commonly employed bioorthogonal reactions include disulfide exchange reactions,8,9 Michael addition with α,β-unsaturated carbonyl compounds,10,11 condensation reactions such as hydrazone or oxime formation,12 Diels–Alder reactions,13,14 click reactions,15–17etc. However, these reactions have several limitations, such as some of the reaction products are sensitive to other nucleophiles such as glutathione present in the milleu,11,18 limited stability at physiological pH,19 requirement of toxic catalysts such as Cu(I)20 or tedious synthesis of strained olefin substrates.21,22 We have recently shown that hydrazone and oxime reactions can be performed at physiological pH using carboxylic acid as a catalyst or under saline conditions.23,24 The hydrolytic stability of such products at physiological pH could also be improved by tuning the electronic characteristics of the hydrazide moiety.25
Reactions involving 1,2-aminothiols are especially interesting because they are naturally present in proteins as N-terminal cysteine. The orthogonal condensation reaction between 1,2-aminothiols and aldehydes to form thiazolidine is an interesting bioconjugation reaction that is poorly explored. Over the years, the thiazolidine chemistry has been explored to develop antibody–drug conjugates,26,27 as a protecting group for N-terminal cysteines,28–31 and for developing cyclic peptides.32,33 In spite of the large body of literature on the 1,2-aminothiol reaction with aldehydes, there has been limited information on the rate of the reaction and stability of the thiazolidine products under physiological conditions.26,27,34–37 It is generally believed that the reaction requires acidic pH26,35,37–40 and a long reaction time,26,27,34–39 and that the thiazolidine product is prone to undergo hydrolysis under physiological conditions.26,27,34,40,41 A lot of research has been dedicated to achieving stable thiazolidine products by designing different substrates. The most common approach is the preparation of aldehydes having a labile ester in the proximity such that the thiazolidine product can undergo ring rearrangement to afford pseudoproline derivatives.34,41,42 Recently, ortho-boronic acid modified benzaldehydes have been prepared to improve the reaction kinetics and to drive the reaction at neutral pH.35,37 Since there are conflicting reports on the reaction conditions and product stability, we decided to examine this reaction carefully using a small molecule model and demonstrate its efficacy to conjugate peptides having N-terminal cysteine with aldehydes containing biomolecules that have therapeutic potential.
To evaluate thiazolidine formation under aqueous conditions, we performed the reaction by mixing L-cysteine with a 16-fold molar excess of propionaldehyde in deuterated phosphate buffer. The 1H-NMR of the reaction mixture showed a fast and complete conversion to the thiazolidine product at pD = 5, 7.4 and 9, as the cysteine resonances were completely disappeared within 5 minutes (Fig. 1 and Fig. S1–S8, ESI†). At pD = 3 thiazolidine formation was considerably slow with complete conversion within 30 minutes, which is presumably due to the less nucleophilic amine/thiol groups (Fig. S9 and Fig. S10, ESI†). These results demonstrate that the reaction between aliphatic aldehydes and 1,2-aminothiols is fast and efficient at a broad range of pH (pH = 5–9) with millimolar concentrations of the substrates. Interestingly, at neutral pD, we observed acidification of the reaction mixture (decrease in pD) as the reaction progressed and therefore an additional base (NaOD) was added to maintain the pD.
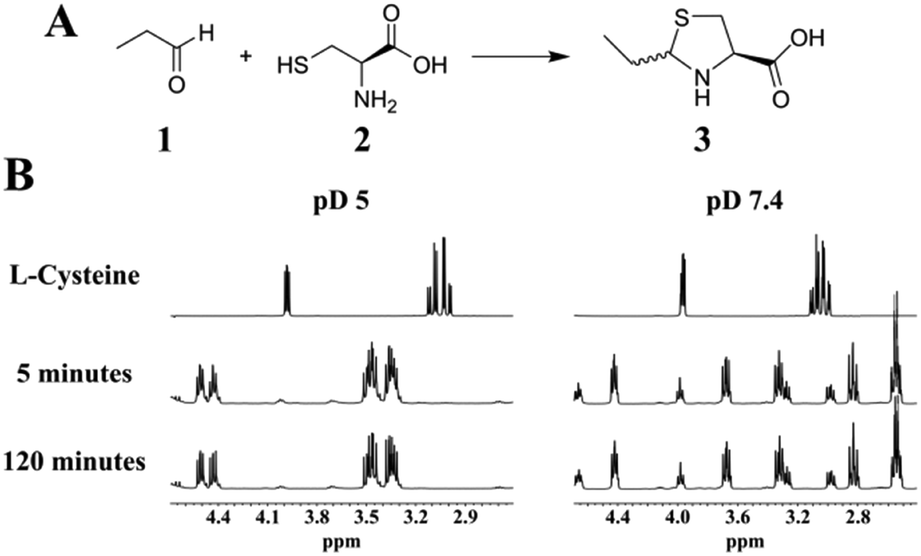 | ||
| Fig. 1 (A) Scheme of the condensation between propionaldehyde and L-cysteine via thiazolidine formation. (B) Representative 1H-NMR spectra at pD 5 and pD 7.4 of the starting material L-cysteine and the reaction mixture recorded after 5 and 120 minutes. | ||
The 1H-NMR analysis of the reaction products revealed different resonances under acidic and neutral conditions. This is presumably due to the deprotonation of the thiazolidine nitrogen that creates a new diastereo center, giving rise to a different splitting pattern of the 1H-NMR spectra at pD 7.4. To test our hypothesis of protonation/deprotonation of the amino group, we performed a pD titration experiment using the purified product (2RS,4R)-2-ethylthiazolidine-4-carboxylic acid (3) and observed the reaction using 1H-NMR analysis at pD 5 and pD 7.4.
These experiments showed that the species underwent rapid interconversion with the change in pD (Fig. 2 and Fig. S11–S13, ESI†), confirming that the structure observed at pD 7.4 is indeed a thiazolidine ring structure. We speculate that the multiple splitting patterns observed in 1H NMR at pD 7.4 led to the belief that thiazolidine products are unstable at physiological pH. As a result there are a limited number of examples in the literature where such reactions have been performed under neutral conditions and it is generally suggested that thiazolidine formation requires acidic conditions and the products are unstable under neutral conditions.26,27,34–42 We believe that the different intensities of the two sets of signals observed at pD 7.4 are due to the interaction of the carboxylic acid with the syn-NH protons that form a zwitterion which makes the deprotonation of the anti-NH proton more favorable. The structure of the thiazolidine ring was systematically characterized using 1D and 2D NMR spectroscopic analyses with the purified product 3 (Fig. S14–S18, ESI†).
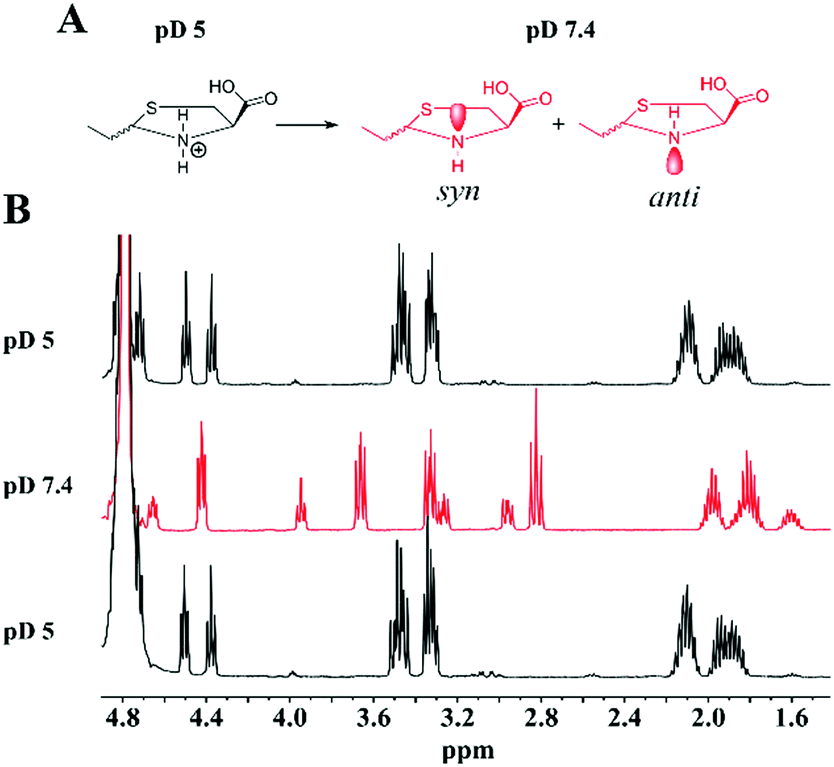 | ||
| Fig. 2 (A) Schematic representation of the protonation/deprotonation of 3 at pD 5 and pD 7.4. (B) Representative 1H-NMR spectra that show the interconversion of the thiazolidine species observed upon pD adjustment. | ||
Once we had established the formation of thiazolidines, we evaluated the stability of the heterocycle at pD 3, 5, 7.4 and 9 using 3 as a model product and examined its integrity periodically via1H-NMR. These studies indicated that the thiazolidine products are extremely stable at the pDs evaluated and revealed no sign of degradation even after 7 days (Fig. S19, ESI†). The efficiency of thiazolidine formation was extended to different aldehyde substrates under neutral conditions. Aliphatic aldehydes, such as butyraldehyde and trimethylacetaldehyde, showed similar results to propionaldehyde as the reaction was completed within 5 minutes (Fig. S20–S23, ESI†). With less reactive aromatic aldehydes, such as benzaldehyde and 4-hydroxybenzaldehyde, the reaction was slower and therefore we calculated the rate of the reaction assuming pseudo-first-order reaction kinetics (Fig. S24 and S25, ESI†). The rate constant observed for benzaldehyde (k1 = 0.0146 min−1) was 4.3-fold higher than that for electron-rich 4-hydroxybenzaldehyde (k1 = 0.0034 min−1). The electron-poor 4-nitrobenzaldehyde could not be studied because it was insoluble under aqueous conditions with 10% deuterated DMSO. The fast formation kinetics of thiazolidine at neutral pH and the high stability of the heterocycle formed suggest that such a reaction could be very advantageous for catalyst-free bioconjugation applications.
Encouraged by the positive results, we explored the potential utility of the thiazolidine conjugation chemistry for peptide ligation reactions. For this purpose, we performed a condensation reaction with a model elastin mimetic peptide (CVGVAPG), which contains an N-terminal cysteine residue and propionaldehyde (Fig. 3). The peptide was mixed with different equivalents of propionaldehyde in phosphate buffer saline (PBS, pH 7.4), keeping the final peptide concentration at 2 mM, and the reaction progress was monitored by HPLC.
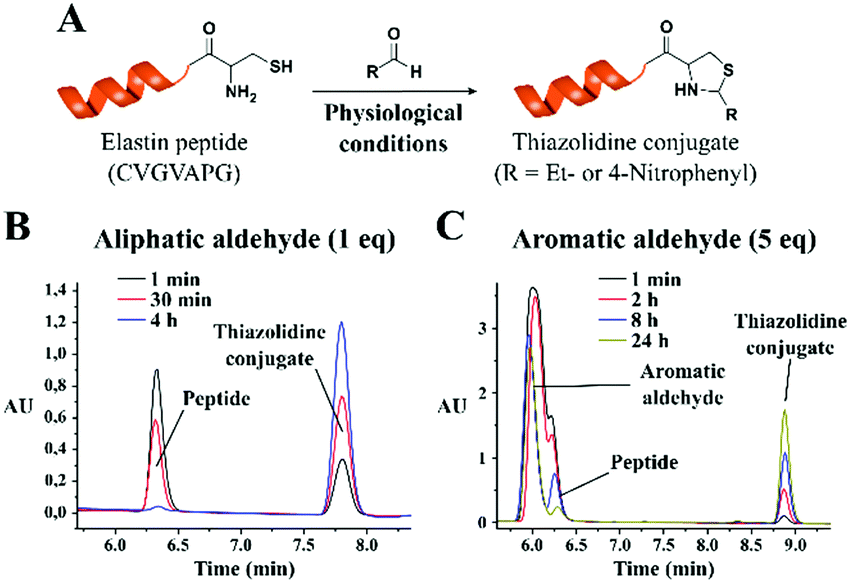 | ||
| Fig. 3 (A) Schematic thiazolidine condensation between aldehydes and elastin mimetic peptide. HPLC chromatogram showing the conjugation reaction with (B) 1 equivalent of propionaldehyde and (C) 5 equivalents of 4-nitrobenzaldehyde at neutral pH. | ||
Similar to what was observed with the small molecule system, the thiazolidine conjugate (retention time 7.8 minutes) was readily formed, and the conjugation was completed in less than 4 h (as the starting material with a retention time of 6.3 minutes disappeared) with only 1 equivalent of propionaldehyde (Fig. 3B). It is worth mentioning that the above reaction was performed in the absence of dithiothreitol (DTT), which resulted in the peptide dimerization due to disulfide formation. When the reaction was performed in the presence of DTT (2 mM final concentration), the reaction was complete within 30 minutes with an equimolar concentration of the peptide and aldehyde group (Fig. S26, ESI†). The kinetics of the reaction could be further improved by increasing the peptide/aldehyde ratio. With 2.5 equivalents of propionaldehyde, we observed a faster reaction rate with 75% conversion within 1 minute (Fig. S27, ESI†). When the propionaldehyde concentration was further increased to 5 equivalents, we observed 85% conversion within 1 minute (Fig. S28, ESI†). When the reaction was performed with an excess of the aldehyde substrate, the formation of disulfide could not be observed, as the reaction rate was extremely fast.
Thereafter we studied the formation of thiazolidines with less reactive aromatic aldehydes using the same peptide and 4-nitrobenzaldehyde as a model substrate. As anticipated, with less reactive aromatic aldehydes we observed a significant decrease in the reaction rate. The reaction was not efficient when the equimolar functionality of aromatic aldehyde was used (27% conversion after 24 hours, Fig. S29, ESI†). However, the reaction rate increased with 5 equivalents of aromatic aldehyde. The analysis of the reaction mixture showed 52% conversion after 8 hours and 87% conversion after 24 hours (Fig. 3C). With 25 equivalents of aldehyde substrate, the reaction was complete within 8 hours (77% conversion after 2 hours, Fig. S30, ESI†). Of note, all these experiments were carried out in the presence of DTT to avoid disulfide formation.
Further, the versatility of this condensation reaction was demonstrated by developing an oligonucleotide–peptide conjugate. As a model peptide, we used the Tat sequence (CYGRKKRRQRRR), which contains an N-terminal cysteine residue and performed the condensation reaction with a model short interfering RNA (siRNA) that contains an aldehyde modification at the 5′-end. The N-terminal cysteine peptide was added in excess (100 equivalents) to ensure the complete conjugation of the aldehyde modified siRNA (10 μM) under physiological conditions. The HPLC chromatogram (monitored at 260 nm for siRNA) of the reaction mixture showed complete conversion to a thiazolidine conjugated siRNA after 30 minutes (Fig. 4).
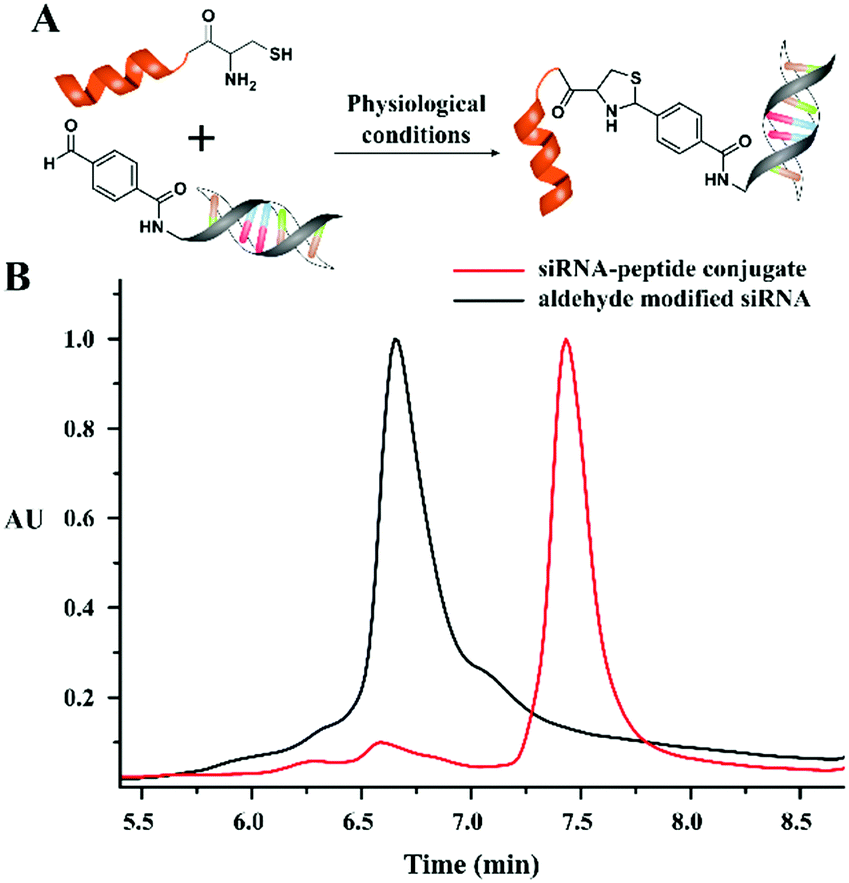 | ||
| Fig. 4 (A) Schematic thiazolidine condensation between aldehyde functionalized siRNA and Tat mimetic peptide. (B) HPLC chromatogram showing the conjugation of the peptide with the aldehyde modified siRNA. | ||
In summary, we demonstrate for the first time that the condensation reaction between 1,2-aminothiols and aliphatic aldehydes is highly specific and fast under physiological conditions. The obtained thiazolidine product was highly stable at both acidic and neutral pH. The efficiency of such a reaction towards chemical ligation was also demonstrated by coupling peptides with small molecule models as well as oligonucleotides. We believe our results will introduce the thiazolidine click reaction to the forefront of bioconjugation reactions and change the misconceptions regarding thiazolidine formation. This study offers a catalyst-free alternative to click chemistry for conjugating sensitive biomolecules.
This research was funded by the European Commission through the Marie Curie Initial Training Network iTERM (Image materials for Tissue Engineering and Regenerative Medicine) Grant agreement no. 607868.
Conflicts of interest
There are no conflicts to declare.Notes and references
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974 CrossRef CAS PubMed.
- C. P. Ramil and Q. Lin, Chem. Commun., 2013, 49, 11007 RSC.
- D. M. Patterson and J. A. Prescher, Curr. Opin. Chem. Biol., 2015, 28, 141 CrossRef CAS PubMed.
- K. E. Beatty, J. C. Liu, F. Xie, D. C. Dieterich, E. M. Schuman, Q. Wang and D. A. Tirrell, Angew. Chem., Int. Ed., 2006, 118, 7524 CrossRef.
- G. U. Nienhaus, Angew. Chem., Int. Ed., 2008, 47, 8992 CrossRef CAS PubMed.
- A. Salic and T. J. Mitchison, PNAS, 2008, 105, 2415 CrossRef CAS PubMed.
- M. Grammel and H. C. Hang, Nat. Chem. Biol., 2013, 9, 475 CrossRef CAS PubMed.
- S. I. van Kasteren, H. B. Kramer, H. H. Jensen, S. J. Campbell, J. Kirkpatrick, N. J. Oldham, D. C. Anthony and B. G. Davis, Nature, 2007, 446, 1105 CrossRef CAS PubMed.
- M. H. Stenzel, ACS Macro Lett., 2013, 2, 14 CrossRef CAS.
- C. M. Cruz, M. Ortega-Muñoz, F. J. López-Jaramillo, F. Hernández-Mateo, V. Blanco and F. Santoyo-González, Adv. Synth. Catal., 2016, 358, 3394 CrossRef CAS.
- M. Z. Chen, N. S. Moily, J. L. Bridgford, R. J. Wood, M. Radwan, T. A. Smith, Z. Song, B. Z. Tang, L. Tilley, X. Xu, G. E. Reid, M. A. Pouladi, Y. Hong and D. M. Hatters, Nat. Commun., 2017, 8, 474 CrossRef PubMed.
- D. K. Kölmel and E. T. Kool, Chem. Rev., 2017, 117, 10358 CrossRef PubMed.
- A. D. de Araújo, J. M. Palomo, J. Cramer, O. Seitz, K. Alexandrov and H. Waldmann, Chem. – Eur. J., 2006, 12, 6095 CrossRef PubMed.
- V. Steven and D. Graham, Org. Biomol. Chem., 2008, 6, 3781 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef.
- Q. Wang, T. R. Chan, R. Hilgraf, V. V. Fokin, K. B. Sharpless and M. G. Finn, J. Am. Chem. Soc., 2003, 125, 3192 CrossRef CAS PubMed.
- C.-H. Jung and J. A. Thomas, Arch. Biochem. Biophys., 1996, 335, 61 CrossRef CAS PubMed.
- J. Kalia and R. T. Raines, Angew. Chem., Int. Ed., 2008, 120, 7633 CrossRef.
- A. E. Speers, G. C. Adam and B. F. Cravatt, J. Am. Chem. Soc., 2003, 125, 4686 CrossRef CAS PubMed.
- J. A. Codelli, J. M. Baskin, N. J. Agard and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 11486 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Org. Lett., 2008, 10, 3097 CrossRef CAS PubMed.
- S. Wang, D. Gurav, O. P. Oommen and O. P. Varghese, Chem. – Eur. J., 2015, 21, 5980 CrossRef CAS PubMed.
- S. Wang, G. N. Nawale, S. Kadekar, O. P. Oommen, N. K. Jena, S. Chakraborty, J. Hilborn and O. P. Varghese, Sci. Rep., 2018, 8, 2193 CrossRef PubMed.
- O. P. Oommen, S. Wang, M. Kisiel, M. Sloff, J. Hilborn and O. P. Varghese, Adv. Funct. Mater., 2013, 23, 1273 CrossRef CAS.
- G. Casi, N. Huguenin-Dezot, K. Zuberbühler, J. Scheuermann and D. Neri, J. Am. Chem. Soc., 2012, 134, 5887 CrossRef CAS PubMed.
- G. J. L. Bernardes, M. Steiner, I. Hartmann, D. Neri and G. Casi, Nat. Protoc., 2013, 8, 2079 CrossRef CAS PubMed.
- I. E. Gentle, D. P. De Souza and M. Baca, Bioconjugate Chem., 2004, 15, 658 CrossRef CAS PubMed.
- D. Bang, B. L. Pentelute and S. B. H. Kent, Angew. Chem., Int. Ed., 2006, 45, 3985 CrossRef CAS PubMed.
- M. Jbara, S. K. Maity, M. Seenaiah and A. Brik, J. Am. Chem. Soc., 2016, 138, 5069 CrossRef CAS PubMed.
- H. Katayama and S. Morisue, Tetrahedron, 2017, 73, 3541 CrossRef CAS.
- P. Botti, T. D. Pallin and J. P. Tam, J. Am. Chem. Soc., 1996, 118, 10018 CrossRef CAS.
- A. Zipperer, M. C. Konnerth, C. Laux, A. Berscheid, D. Janek, C. Weidenmaier, M. Burian, N. A. Schilling, C. Slavetinsky, M. Marschal, M. Willmann, H. Kalbacher, B. Schittek, H. Brötz-Oesterhelt, S. Grond, A. Peschel and B. Krismer, Nature, 2016, 535, 511 CrossRef CAS PubMed.
- A. M. Oelker, J. A. Berlin, M. Wathier and M. W. Grinstaff, Biomacromolecules, 2011, 12, 1658 CrossRef CAS PubMed.
- A. Bandyopadhyay, S. Cambray and J. Gao, Chem. Sci., 2016, 7, 4589 RSC.
- X. Bi, K. K. Pasunooti, A. H. Tareq, J. Takyi-Williams and C.-F. Liu, Org. Biomol. Chem., 2016, 14, 5282 RSC.
- H. Faustino, M. J. S. A. Silva, L. F. Veiros, G. J. L. Bernardes and P. M. P. Gois, Chem. Sci., 2016, 7, 5052 RSC.
- J. Shao and J. P. Tam, J. Am. Chem. Soc., 1995, 117, 3893 CrossRef CAS.
- L. Zhang and J. P. Tam, Anal. Biochem., 1996, 233, 87 CrossRef CAS PubMed.
- M. Jbara, S. Laps, S. K. Maity and A. Brik, Chem. – Eur. J., 2016, 22, 14851 CrossRef PubMed.
- M. Wathier, C. S. Johnson, T. Kim and M. W. Grinstaff, Bioconjugate Chem., 2006, 17, 873 CrossRef CAS PubMed.
- C. F. Liu and J. P. Tam, PNAS, 1994, 91, 6584 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopic and analytical data. See DOI: 10.1039/c8cc05405c |
| This journal is © The Royal Society of Chemistry 2018 |