
Actinide-transition metal bonding in heterobimetallic uranium– and thorium–molybdenum paddlewheel complexes†

Abstract
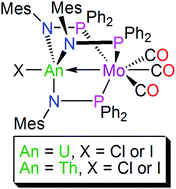
We report the preparation of four heterobimetallic uranium– and thorium–molybdenum paddlewheel complexes. The characterisation data suggest the presence of Mo → An σ-interactions in all cases. These complexes represent unprecedented actinide-group 6 metal–metal bonds, where before heterobimetallic uranium–metal bonds were restricted to group 7–11 metals.
- This article is part of the themed collections: Celebrating our 2020 Prize and Award winners and New molecules and materials from the f-block
 Please wait while we load your content...
Please wait while we load your content...

