
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Essential but sparse collagen hydroxylysyl post-translational modifications detected by DNP NMR†
Wing Ying
Chow
 a,
Rui
Li
b,
Ieva
Goldberga
b,
David G.
Reid
b,
Rakesh
Rajan
b,
Jonathan
Clark
c,
Hartmut
Oschkinat
*a,
Melinda J.
Duer
*b,
Robert
Hayward
d and
Catherine M.
Shanahan
d
a,
Rui
Li
b,
Ieva
Goldberga
b,
David G.
Reid
b,
Rakesh
Rajan
b,
Jonathan
Clark
c,
Hartmut
Oschkinat
*a,
Melinda J.
Duer
*b,
Robert
Hayward
d and
Catherine M.
Shanahan
d
aLeibniz Forschungsinstitut für Molekulare Pharmakologie, Campus Buch, Robert-Roessle Str. 10, Berlin 13125, Germany. E-mail: oschkinat@fmp-berlin.de; Tel: +49 30 94793 160
bDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: mjd13@cam.ac.uk; Fax: +44 1223 336362; Tel: +44 1223 736394
cBabraham Institute, Babraham Research Campus, Cambridge CB22 3AT, UK
dBHF Centre of Research Excellence, Cardiovascular Division, King's College London, London SE5 9NU, UK
First published on 5th October 2018
Abstract
The sparse but functionally essential post-translational collagen modification 5-hydroxylysine can undergo further transformations, including crosslinking, O-glycosylation, and glycation. Dynamic nuclear polarization (DNP) and stable isotope enriched lysine incorporation provide sufficient solid-state NMR sensitivity to identify these adducts directly in skin and vascular smooth muscle cell extracellular matrix (ECM), without extraction procedures, by comparison with chemical shifts of model compounds. Thus, DNP provides access to the elucidation of structural consequences of collagen modifications in intact tissue.
Lysine modifications are critical to collagen ECM structure, function, and properties.1,2 These usually start with hydroxylation at the δ-(i.e. the 5-) carbon and lead to a cascade of reactions that generate diverse chemical structures. The modifications can be enzymatically-driven as part of collagen and ECM biosynthesis, but can also occur without enzymatic involvement in conditions such as diabetes, cancer and ageing. Interruption of the lysine modification process may lead to embryonic and perinatal lethality in mouse, and osteogenesis imperfecta and Ehler–Danlos disease in humans.1,3 Being able to observe such lysine modifications in tissues without the application of extraction procedures is critical for understanding their physiological necessity, as well as the role they play in pathological and degradation processes.
Lysyl hydroxylases4 modify Cδ in less than 2%5 of lysine residues in collagen to yield (R)-5-hydroxylysine (Hyl). Hyl residues are critical sites for further modifications, including their attachment to other Lys residues in a variety of cross-links,6,7 (ESI,† Schemes S1 and S2), adduction of sugar species via specific and essential enzymatic glycosylation,8,9 (ESI,† Scheme S3), and invariably deleterious spontaneously occurring glycation10,11 (ESI,† Scheme S4). The generation of Hyl and all three types of subsequent modifications will produce broadly predictable changes in the NMR spectra of ECM materials; the challenge is detecting these low abundance species against an overwhelming background of irrelevant signals.
Conventional analysis of collagen modifications relies on mass spectrometry.12–14 The identification procedure demands many, often chemical, pre-treatments.15,16 While sensitive and potentially quantitative, these approaches are destructive and raise the possibility of side reactions or degradation during processing. There is a need to develop complementary analytical techniques to arrive at a full picture of the identity and quantity of collagen modifications in a range of biological samples.
Solid-state NMR (ssNMR) offers the possibility of identifying such collagen modifications in intact tissue without extensive processing or purification. With ssNMR, the types of species present may be identified, and valuable structural and dynamical information on crosslinked, glycosylated and glycated sites within intact ECM may be obtained. A significant challenge in this approach is the relatively low sensitivity of solid-state NMR spectroscopy, exacerbated by the low natural abundance of NMR-active nuclei and the fact that the residues of interest occur at levels well under 2% of all residues found in collagen proteins.
The strategy applied here combines stable isotopic labelling with NMR signal enhancement by DNP.17–19 To show the wide applicability of this strategy, two different kinds of samples were investigated: skin obtained from mice fed a (U-13C6)-lysine diet, and ECM generated by vascular smooth muscle cell (VSMC) culture as a potential in vitro model with relevance to vascular diseases. Labelling with NMR-active stable isotopes (e.g.13C, 15N) is a well-established procedure for enhancing sensitivity and information content.20,21 We have developed cell culture methods for generating labelled ECM by supplementing growth media with isotope-enriched amino acids.22 A dietary approach is particularly effective for labelling mice in essential amino acids like lysine, where incorporation from diet is not in competition with de novo native biosynthesis.23 Accordingly, 13C-enriched murine skin was obtained by direct feeding of mice with (U-13C6)-lysine, while VSMC ECM24 was labelled using growth medium supplemented with (U-13C6,15N2)-lysine. Note that the dietary approach does not completely isotope enrich the experimental animal as tissue biosynthesis and turnover can vary; we achieved 45% 13C-lysine enrichment on mouse skin.
The second component of our strategy to overcome the significant challenge of observing dilute molecular sites was to use DNP25 to enhance signal sensitivity further. Despite the inhomogeneous signal broadening inevitable at cryogenic temperatures due to reduction of motional averaging, and the homogeneous signal broadening due to the presence of the radicals, the increased sensitivity from DNP enhancement renders weak signals observable that do not emerge above noise within a reasonable timeframe by conventional NMR. DNP has been usefully applied to large molecular systems, such as ribosomes,26 and assembled HIV capsid proteins.27 In a similar vein, we use DNP signal enhancement to probe sparse modifications of specifically labelled residues within intact collagen fibrils: large well-ordered, super-coiled, cross-linked units of triple-helical collagen, each around 300 kDa in molecular weight.
The 13C-Lys enriched samples are dry, solid biomaterials, hence we doped 10–15 mg of each with 17–20 mM AMUPol28 dissolved in water (75% D2O, 25% H2O), centrifuged the added radical solution several times in the rotor, and incubated the rotor in the fridge. This procedure resembles the incipient wetness impregnation technique as applied to porous materials.29 In contrast to common practice in DNP-NMR, glycerol was not added as it modifies collagen stability.30 Mouse skin samples do exhibit up to 50% higher enhancements with glycerol (ESI,† Fig. S1), however, the VSMC ECM sample without glycerol showed higher enhancement than the glycerol-containing mouse skin sample. Furthermore, glycerol addition leads to artefacts in the same region as the Hyl signals (ESI,† Fig. S2). Considering the potential structural complications, and as Hyl signals can already be observed in both samples without glycerol, we omitted it for the rest of this study.
DNP enhancements of 26.5 ± 1.5 for skin and 57 ± 4 for VSMC ECM were measured by comparing microwave-on versus microwave-off 1D 13C MAS NMR spectra. The error is obtained by calculating the average enhancement of the most (generally Cα) and the least (generally Cβ/Cγ) enhanced signals (ESI,† Fig. S1), indicating uniform DNP enhancements. Taking possible depolarization effects of up to 60% into account,31 these enhancements represent one to two orders of magnitude reduction in NMR signal averaging time. All experiments were carried out at 110 K, the lowest temperature where we have the required long-term stability to complete the NMR experiments.
As expected, the 1D 13C CP DNP NMR spectra of the isotope enriched skin and VSMC ECM are dominated by signals from unmodified Lys in collagen32 and other proteins (ESI,† Fig. S2). Cross peaks in 2D dipolar assisted rotational resonance (DARR)33 spectra (Fig. 1A and ESI,† Fig. S2, S3) reveal proximal 13C–13C pairs. While the most intense signals arise from Lys residues, three distinct Hyl-derived spin systems can be resolved in the DARR spectra, highlighted in Fig. S2 (ESI†) (red boxes) and detailed in Fig. 1A. As these signals can only arise from derivatives of 13C-enriched Lys, they must correspond to Lys post-translational modifications.
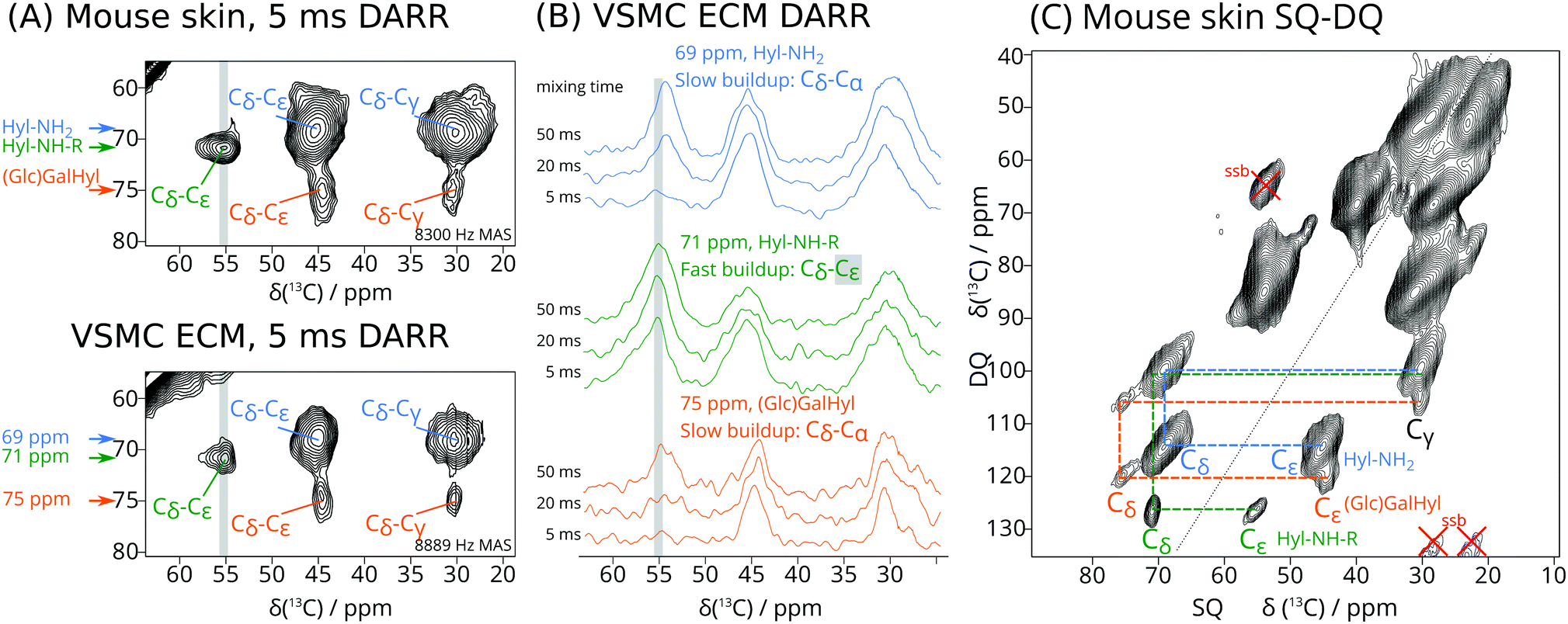 | ||
| Fig. 1 (A) Regions of 2D 13C–13C DARR DNP-NMR spectra (5 ms mixing time, 110 K) of U-13C6 Lys labelled skin (15 mg, 8300 Hz MAS, acquisition 17 h, top) and VSMC ECM (15 mg, 8889 Hz MAS, acquisition time 7 h, bottom) showing cross peaks between Hyl Cδ and other 13C-labelled carbons in Hyl and its derivatives. Blue annotations at 69 ppm (F1) depict underivatized Hyl (Hyl-NH2), green annotations at 71 ppm (F1) modified Hyl likely due to crosslinks and/or glycation (Hyl-NH-R), and orange annotations at 75 ppm (F1) glycosylated Hyl ((Glc)GalHyl). (B) Traces taken from DARR correlations of labelled VSMC ECM at different DARR mixing times, showing intensity changes of cross peaks involving Hyl Cδ. The rapid build-up of the correlation with the Cε signal of Hyl-NH-R (green traces) at 56 ppm indicates that this signal can be distinguished from the correlation to the more distant Hyl Cα at 54 ppm, which builds up more slowly. (C) 2D 13C–13C SQ-DQ DNP NMR spectrum of labelled skin, showing the same Hyl Cδ–Cε signal that we have identified in (A) and (B). | ||
Specifically, the cross peaks in Fig. 1A are consistent with Hyl and its derivatives, with the signals of the hydroxylated Cδ around 70 ppm. The largest signals, centred at 69 ppm, likely correspond to Hyl residues that have not undergone further modification and show good agreement with solution-state NMR chemical shifts (see ESI†), thus we refer to this set of signals as Hyl-NH2. A set of weaker but distinct signals at 75 ppm is consistent with O-glycosylated Hyl, in line with solution-state NMR shifts of GalHyl34,35 and GlcGalHyl,35,36 thus we refer to it as (Glc)Gal-Hyl (ESI,† Scheme S3), though we note that we are unable to determine whether Hyl is glycosylated with GlcGal or only Gal at this stage. We expect these signals to be weaker since only up to one third of all Hyl is glycosylated. The well-resolved cross peak at 71 ppm (F1) and 56 ppm (F2) is consistent with a Cδ–Cε cross peak for Hyl derivatized at Nζ (see ESI† for reference model structures), which we term Hyl-NH-R. The rapid build-up of the Hyl-NH-R cross peak at 71–56 ppm (Fig. 1B green traces) distinguishes it from the overlapping but more slowly increasing signal corresponding to the Hyl-NH2 Cδ–Cα correlation, for which the cross peak occurs at 69 ppm F1 and 54 ppm F2 (Fig. 1B blue traces). This effect is especially apparent in the traces from spectra recorded with DARR mixing times of 5 ms (Fig. 1B). From this result, we can deduce that the 71–56 ppm cross peak arises from two carbons that are directly bonded to each other. The signals around 70 ppm are due to Hyl Cδ, thus the 56 ppm signal is identified as Cε by elimination. The SQ-DQ spectrum (Fig. 1C), where one-bond correlations dominate, provides further evidence of this pair of atoms, where we observed the 71–56 ppm correlation occurring at the expected DQ (sum) frequency of 127 ppm.
For Hyl without further modifications (Hyl-NH2), Cε occurs at 45 ppm. However, we can expect that substitutions at the Hyl Nζ primary amino group would shift the Cε signal about 10 ppm to higher frequency, thus providing a rationale for identifying the 56 ppm signal as Cε of Hyl-NH-R. Therefore, the Hyl-NH-R signal may represent Hyl modifications such as early glycation adducts, for instance with the C-1 of 1-deoxyfructose derived from glucose (ESI,† Scheme S4). It may also correspond to enzymatically derived ketoamine crosslinks (e.g. HLKNL in ESI,† Scheme S2). Chemical shifts in Table 1 are consistent with those of appropriate model compounds.34–36 While the conventional aldimine structural formulation of immature aldimine cross links (e.g. deH-HLNL in ESI,† Scheme S2) predicts a signal at 160–165 ppm for the ε-imino carbon derived U-13C6 allysine, such signals were not observed.
| PTM | Cα | Cβ | Cγ | Cδ | Cε | %Skin | %VSMC |
|---|---|---|---|---|---|---|---|
| Lys | 54 | 30 | 23 | 28 | 40 | 100 | 100 |
| Hyl (Hyl-NH2) | 54 | 28 | 30 | 69 | 45 | 14 | 7 |
| Cross links/glycation (Hyl-NH-R) | 54 | 30 | 28 | 71 | 56 | 3 | 4 |
| (Glc)GalHyl | 54 | 28 | 30 | 75 | 44 | 3 | 2 |
Independent evidence that 13C signals around 70 ppm are due to Hyl Cδ and its derivatives comes from heteronuclear correlations involving Hyl 15Nζ in the (U-13C6, 15N2)-Lys enriched VSMC (Fig. 2 and Fig. S4, ESI†). Fig. 2 shows the region centred at 38 ppm 15N chemical shift corresponding to the Lys or Hyl 6-amino (Nζ) group. The magnetisation evolved on 15N (Lys/Hyl Nζ) was transferred to neighbouring Cε, and then further down the carbon skeleton by DARR mixing to the hydroxylated Cδ around 70 ppm 13C chemical shift, resulting in a correlation connecting Hyl Nζ (38 ppm) to the corresponding Cδ (69–71 ppm).
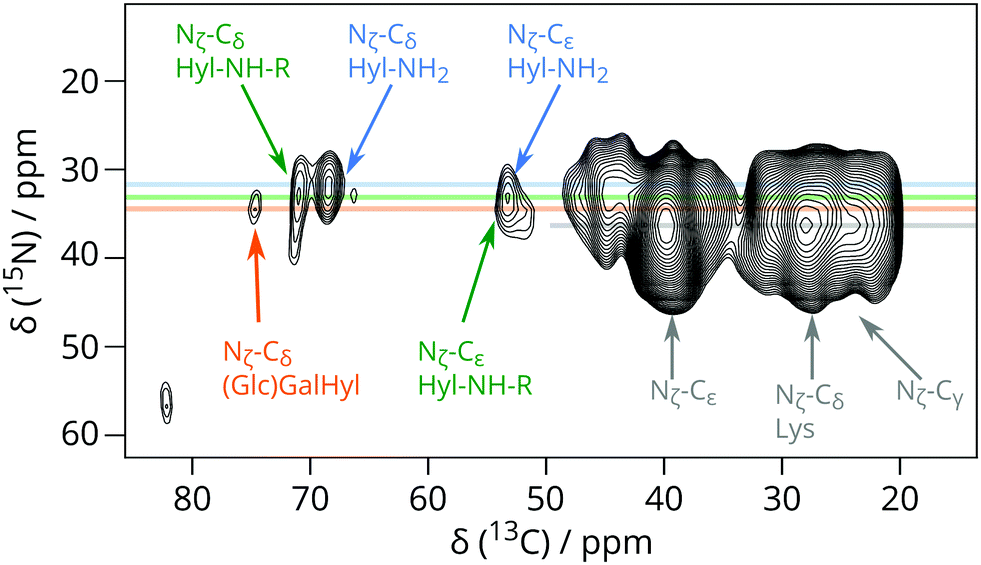 | ||
| Fig. 2 Detail from 15N–13C correlation DNP-NMR experiment showing the correlation between Hyl Cδ and Hyl Nζ signals for (U-13C6, 15N2)-Lys enriched VSMC. This experiment took 51 hours. | ||
In summary, combining Lys isotope labelling and DNP enhancement enabled solid-state NMR observation of Hyl and other post-translational modifications in freeze-dried but otherwise intact collagenous ECM. We identified those due to O-glycosylated Hyl and signals indicative of N-adducts that correspond to early glycation products and/or crosslinks. These were present at levels of only a few percent of total Lys; we have thus conclusively shown that detection of these low abundance species within biologically-produced materials is possible without substantial extraction or purification. Our study attests to the potential of combining specific labelling and DNP to study the structural roles of Hyl in more complex organic–inorganic biocomposites such as bone22,37 and pathological vascular calcifications.38 The similarity of Hyl and derivatives between mammalian VSMC ECM generated by cell culture and native mouse skin supports our use of in vitro biomaterials as convenient and ethical model systems amenable to isotope labelling, in which the post-translational modification machinery is functioning faithfully. We are currently identifying the species adducted to Hyl using 13C-enriched sugar precursors, and increasing the proportions of 13C-labelled Lys and Hyl in our ECM to characterize cross links in more detail and under biologically relevant conditions. We envisage that such NMR-based techniques will be central in understanding the structural roles of Hyl and its modifications at the atomic level, to further our understanding of their physiological necessity in healthy tissue, and the part they play in disease and ageing.
Funding from the U.K. Medical Research Council for DGR and RR (grant RG75828), the SENS Foundation for JC, the Engineering and Physical Sciences Research Council for PhD studentships for RL and IG, postdoctoral fellowship funding from the DAAD and Leibniz Association for WYC, the CSC Cambridge Scholarship for PhD studentship for RL, and support by iNEXT, grant number 653706, funded by the Horizon 2020 programme of the European Commission.
Conflicts of interest
There are no conflicts of interest. Animal procedures complied with institutional ethical guidelines, and the U.K. Animals (Scientific Procedures) Act 1986.Notes and references
- J. Myllyharju and K. I. Kivirikko, Trends Genet., 2004, 20, 33 CrossRef CAS PubMed.
- K. Takaluoma, M. Hyry, J. Lantto, R. Sormunen, R. A. Bank, K. I. Kivirikko, J. Myllyharju and R. Soininen, J. Biol. Chem., 2007, 282, 6588 CrossRef CAS PubMed.
- K. Rautavuoma, K. Takaluoma, R. Sormunen, J. Myllyharju, K. I. Kivirikko and R. Soininen, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14120 CrossRef CAS PubMed.
- M. Yamauchi and M. Sricholpech, Essays Biochem., 2012, 52, 113 CrossRef CAS PubMed.
- J. Myllyharju, Collagen, 2005, 247, 115 CAS.
- D. R. Eyre, M. A. Paz and P. M. Gallop, Annu. Rev. Biochem., 1984, 53, 717 CrossRef CAS PubMed.
- D. R. Eyre, I. R. Dickson and K. Van Ness, Biochem. J., 1988, 252, 495 CrossRef CAS PubMed.
- M. Sricholpech, I. Perdivara, M. Yokoyama, H. Nagaoka, M. Terajima, K. B. Tomer and M. Yamauchi, J. Biol. Chem., 2012, 287, 22998 CrossRef CAS PubMed.
- M. Terajima, I. Perdivara, M. Sricholpech, Y. Deguchi, N. Pleshko, K. B. Tomer and M. Yamauchi, J. Biol. Chem., 2014, 289, 22636 CrossRef CAS PubMed.
- A. G. Huebschmann, J. G. Regensteiner, H. Vlassara and J. E. B. Reusch, Diabetes Care, 2006, 29, 1420 CrossRef CAS PubMed.
- V. M. Monnier, G. T. Mustata, K. L. Biemel, O. Reihl, M. O. Lederer, Z. Y. Dai and D. R. Sell, Ann. N. Y. Acad. Sci., 2005, 533 CrossRef CAS PubMed.
- A. Naba, O. M. T. Pearce, A. Del Rosario, D. Ma, H. Ding, V. Rajeeve, P. R. Cutillas, F. R. Balkwill and R. O. Hynes, J. Proteome Res., 2017, 16, 3083 CrossRef CAS PubMed.
- W. Y. Jiang, B. Y. Zhou, G. L. Yu, H. Liu, J. B. Zeng, Q. D. Lin, H. L. Xi and H. Liang, Biochem. Genet., 2012, 50, 34 CrossRef CAS PubMed.
- I. Perdivara, M. Yamauchi and K. B. Tomer, Aust. J. Chem., 2013, 66, 760 CrossRef CAS PubMed.
- A. Kondo, O. Ishikawa, K. Okada, Y. Miyachi, S. Abe and Y. Kuboki, Anal. Biochem., 1997, 252, 255 CrossRef CAS PubMed.
- T. J. Sims, N. C. Avery and A. J. Bailey, Methods Mol. Biol., 2000, 139, 11 CAS.
- K. K. Frederick, V. K. Michaelis, M. A. Caporini, L. B. Andreas, G. T. Debelouchina, R. G. Griffin and S. Lindquist, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3642 CrossRef CAS PubMed.
- G. T. Debelouchina, M. J. Bayro, A. W. Fitzpatrick, V. Ladizhansky, M. T. Colvin, M. A. Caporini, C. P. Jaroniec, V. S. Bajaj, M. Rosay, C. E. Macphee, M. Vendruscolo, W. E. Maas, C. M. Dobson and R. G. Griffin, J. Am. Chem. Soc., 2013, 135, 19237 CrossRef CAS PubMed.
- I. V. Sergeyev, L. A. Day, A. Goldbourt and A. E. McDermott, J. Am. Chem. Soc., 2011, 133, 20208 CrossRef CAS PubMed.
- D. M. LeMaster, Prog. Nucl. Magn. Reson. Spectrosc., 1994, 26, 371 CrossRef CAS.
- J.-P. Demers, P. Fricke, C. Shi, V. Chevelkov and A. Lange, Prog. Nucl. Magn. Reson. Spectrosc., 2018, 109, 51 CrossRef CAS.
- W. Y. Chow, R. Rajan, K. H. Muller, D. G. Reid, J. N. Skepper, W. C. Wong, R. A. Brooks, M. Green, D. Bihan, R. W. Farndale, D. A. Slatter, C. M. Shanahan and M. J. Duer, Science, 2014, 344, 742 CrossRef CAS PubMed.
- V. W. C. Wong, D. G. Reid, W. Y. Chow, R. Rajan, M. Green, R. A. Brooks and M. J. Duer, J. Biomol. NMR, 2015, 63, 119 CrossRef CAS PubMed.
- R. Li, R. Rajan, W. C. V. Wong, D. G. Reid, M. J. Duer, V. J. Somovilla, N. Martinez-Saez, G. J. L. Bernardes, R. Hayward and C. M. Shanahan, Chem. Commun., 2017, 53, 13316 RSC.
- A. S. L. Thankamony, J. J. Wittmann, M. Kaushik and B. Corzilius, Prog. Nucl. Magn. Reson. Spectrosc., 2017, 102–103, 120 CrossRef PubMed.
- S. Lange, W. T. Franks, N. Rajagopalan, K. Doring, M. A. Geiger, A. Linden, B. J. van Rossum, G. Kramer, B. Bukau and H. Oschkinat, Sci. Adv., 2016, 2, e1600379 Search PubMed.
- R. Gupta, M. Lu, G. Hou, M. A. Caporini, M. Rosay, W. Maas, J. Struppe, C. Suiter, J. Ahn, I. J. Byeon, W. T. Franks, M. Orwick-Rydmark, A. Bertarello, H. Oschkinat, A. Lesage, G. Pintacuda, A. M. Gronenborn and T. Polenova, J. Phys. Chem. B, 2016, 120, 329 CrossRef CAS PubMed.
- C. Sauvee, M. Rosay, G. Casano, F. Aussenac, R. T. Weber, O. Ouari and P. Tordo, Angew. Chem., Int. Ed., 2013, 52, 10858 CrossRef CAS PubMed.
- A. Lesage, M. Lelli, D. Gajan, M. A. Caporini, V. Vitzthum, P. Mieville, J. Alauzun, A. Roussey, C. Thieuleux, A. Mehdi, G. Bodenhausen, C. Coperet and L. Emsley, J. Am. Chem. Soc., 2010, 132, 15459 CrossRef CAS PubMed.
- G. C. Na, Biochemistry, 1986, 25, 967 CrossRef CAS PubMed.
- F. Mentink-Vigier, S. Paul, D. Lee, A. Feintuch, S. Hediger, S. Vega and G. De Paepe, Phys. Chem. Chem. Phys., 2015, 17, 21824 RSC.
- O. Nikel, D. Laurencin, S. A. McCallum, C. M. Gundberg and D. Vashishth, Langmuir, 2013, 29, 13873 CrossRef CAS PubMed.
- K. Takegoshi, S. Nakamura and T. Terao, Chem. Phys. Lett., 2001, 344, 631 CrossRef CAS.
- P. Lamosa, L. O. Martins, M. S. Da Costa and H. Santos, Appl. Environ. Microbiol., 1998, 64, 3591 CAS.
- P. Allevi, R. Paroni, A. Ragusa and M. Anastasia, Tetrahedron: Asymmetry, 2004, 15, 3139 CrossRef CAS.
- P. Allevi, M. Anastasia, R. Paroni and A. Ragusa, Bioorg. Med. Chem. Lett., 2004, 14, 3319 CrossRef CAS PubMed.
- P. Zhu, J. Xu, N. Sahar, M. D. Morris, D. H. Kohn and A. Ramamoorthy, J. Am. Chem. Soc., 2009, 131, 17064 CrossRef CAS PubMed.
- M. J. Duer, T. Friscic, D. Proudfoot, D. G. Reid, M. Schoppet, C. M. Shanahan, J. N. Skepper and E. R. Wise, Arterioscler., Thromb., Vasc. Biol., 2008, 28, 2030 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc04960b |
| This journal is © The Royal Society of Chemistry 2018 |