
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment and cellular application of visible-light-controllable HNO releasers based on caged Piloty's acid†
Mitsuyasu
Kawaguchi
,
Takuma
Tani
,
Ryoma
Hombu
,
Naoya
Ieda
and
Hidehiko
Nakagawa
 *
*
Graduate School of Pharmaceutical Sciences, Nagoya City University, 3-1 Tanabe-dori, Mizuho-ku, Nagoya, Aichi 467-8603, Japan. E-mail: deco@phar.nagoya-cu.ac.jp
First published on 28th August 2018
Abstract
Nitroxyl (HNO) is a gaseous molecule with unique pharmacological functions distinct from those of nitric oxide. Because HNO is highly reactive with biological molecules, spatiotemporally controllable HNO releasers are required. Herein, we report the first visible-light-controllable HNO releasers, based on caged Piloty's acid, and we demonstrate their applicability in living cells.
Nitroxyl (HNO), a type of reactive nitrogen species (RNS) and the one-electron-reduced form of nitric oxide, was reported by Angeli et al. in the latter half of the 19th century.1 In recent years, HNO has attracted attention due to its unique pharmacological activity, such as enhancement of cardiac contractility,2 and it is considered a therapeutic candidate for cardiovascular disease and cancer.3 However, the precise roles of HNO in vivo are still not well understood, because HNO has very high reactivity with biological thiols (k = 7.6 × 106 M−1 s−1),4 phosphine (k = 8.4 × 106 M−1 s−1) and metal-containing proteins,5 and spontaneously dimerizes to generate N2O (k = 8 × 106 M−1 s−1) in aqueous solution.6 Many HNO donors, such as Angeli's salt (Na2N2O3, AS),7 Piloty's acid derivatives,8 diazeniumdiolates (NONOates),9 acyl nitroso compounds,10 and acyloxy nitroso compounds11 have been developed and used in biological studies, but they all decompose spontaneously to release HNO under physiological conditions. In order to control HNO release more precisely, photocontrollable HNO donors have been developed by our group based on a retro Diels–Alder reaction12 and by Sampson's group based on N-alkoxysulfonamide bearing a O-(3-hydroxy-2-naphthalenyl)-methyl (HNM) photolabile protecting group (PPG).13 However, these compounds are not suitable for cellular experiments, because HNO release is triggered by ultraviolet (UV) irradiation, which is cytotoxic.
Herein, we aimed to develop HNO donors that are photocontrollable by irradiation in the visible light range, that would be applicable in living cells. For this purpose, we focused on Piloty's acid, which releases HNO via deprotonation of its hydroxyl group under basic conditions (Scheme 1a).8 Furthermore, its derivatives, 2-nitro-N-hydroxybenzensulfonamide (compound A) and 2-bromo-N-hydroxybenzensulfonamide (compound B) release HNO even under physiological conditions (pH ≃ 7.4) (Scheme 1b).8c Based on these reports, we designed and synthesized compounds 8 and 9, in which the hydroxyl group is protected by a (7-diethylaminocoumarin-4-yl)methyl group, a kind of PPG releasable by visible light near 400 nm (Scheme 1c).14 By using an HNO selective fluorescence probe, P-Rhod,15 we show that these HNO donors enable visible-light-controllable HNO release in living cells without cytotoxicity.
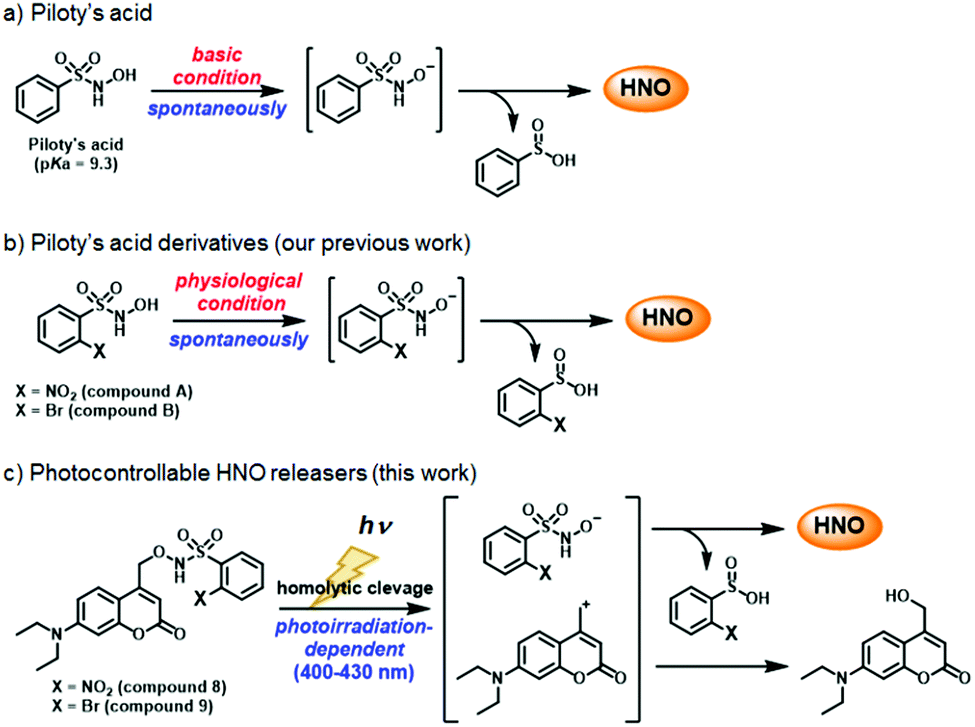 | ||
| Scheme 1 (a) Piloty's acid spontaneously releases HNO under basic conditions. (b) Piloty's acid derivatives bearing –NO2 (compound A) or –Br (compound B) at the ortho position can spontaneously release HNO under physiological neutral conditions due to their low pKa values compared to Piloty's acid. (c) Photocontrollable HNO releasers (8, 9) release HNO in a photoirradiation-dependent manner in the visible light region (400–430 nm) under physiological conditions. | ||
Compounds 8 and 9 were synthesized as shown in Scheme 2. Briefly, Riley oxidation and hydride reduction of 7-diethyl-amino-4-methylcoumarin (1) gave the hydroxymethyl derivative (3) in two steps. Next, (7-diethylaminocoumarin-4-yl)methyl bromide (5) was synthesized by mesylation and bromination of compound 3. SN2 reaction converted 5 to Boc-protected aminooxylated compound 6, which was deprotected with HCl to yield 7. Condensation reaction with benzenesulfonyl chloride derivatives afforded compounds 8 and 9, whose structures were confirmed by NMR, MS, and elemental analysis.
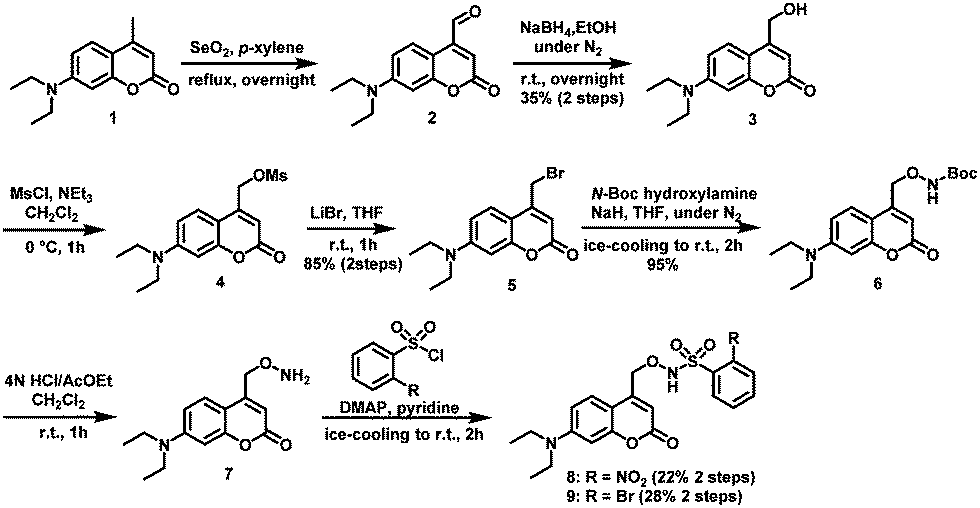 | ||
| Scheme 2 Synthesis of HNO releaser compounds 8 and 9. | ||
The UV-vis spectrum of a solution of compound 8 or 9 in MilliQ water showed strong absorption around 400 nm derived from the coumarin moiety (Fig. S1, ESI†). Therefore, we decided to use a Xe lamp source equipped with a 400–430 nm band pass filter for photodecomposition.
Next, we examined the photodecomposition reaction of compound 8 by means of HPLC (Fig. 1a–e). After photoirradiation, compound 8 (tR = 17.9 min) completely disappeared and new peaks were detected. Among them, two were assigned to 2-nitrobenzene sulfinic acid (compound 11) (tR = 2.8 min) and 7-diethylamino-4-hydroxymethylcoumarin (compound 3) (tR = 7.6 min); these were expected to be generated after HNO release. However, the peak of compound 3 was much smaller than expected, and an unexpected peak (tR = 12.5 min) was observed as the main peak. We purified the eluate corresponding to this peak, and identified the product as 7-diethylaminocoumarin-4-carbaldehyde oxime (compound 12) by examination of the NMR spectrum. We also separately synthesized compound 12 and confirmed that the authentic compound was identical with the photodecomposition product on the basis of NMR and HPLC analyses (Scheme S1 and Fig. S2, ESI†). These results suggest that the main photodecomposition mechanism is not the expected one (path I). Instead, we propose a mechanism (path II) in which compound 12 is mainly generated (Fig. 1f). After photoirradiation, the C–O bond at the coumarinyl 4-position is decomposed by heterolytic cleavage (or homolytic cleavage followed by one electron transfer) and a new C–N bond is formed by nucleophilic attack of an adjacent nitrogen atom. Then sulfinic acid, compound 11, is detached and compound 12 is formed through tautomerization of the nitroso compound. We estimated a fraction of path I from the result of Fig. 1, whose path releases HNO, and found that 7.2% of compound 8 underwent path I reaction (Fig. S3, ESI†).
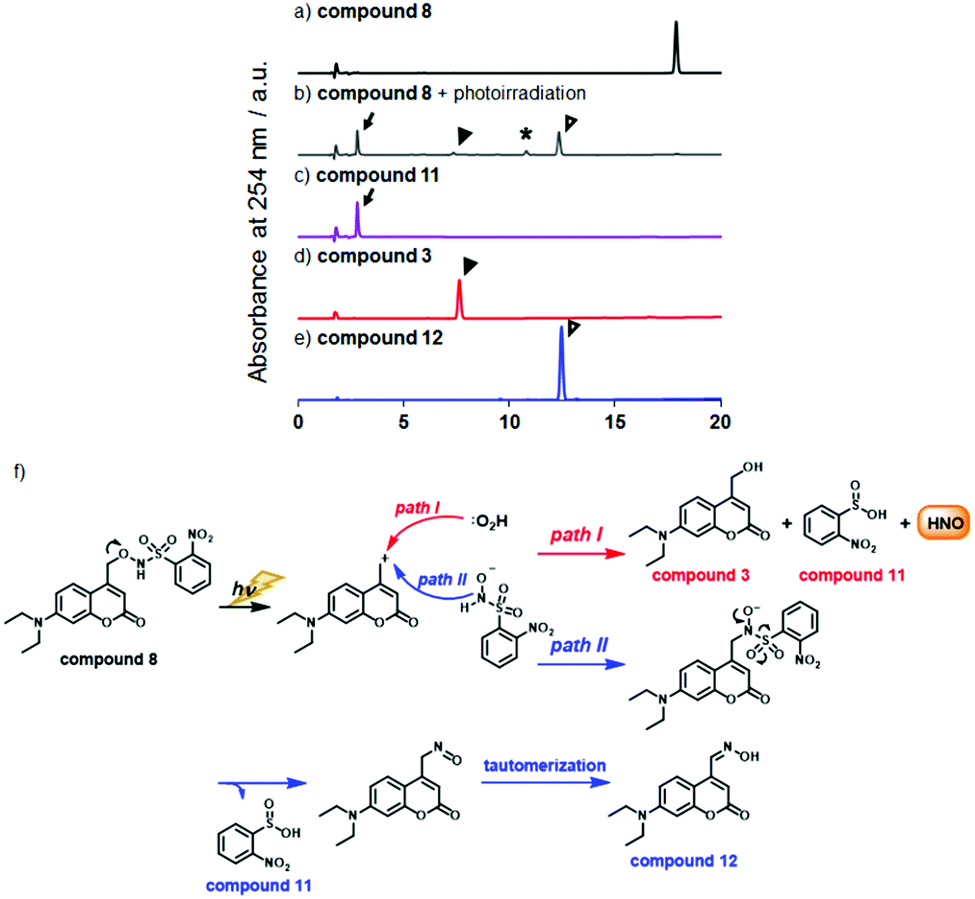 | ||
Fig. 1 Photodecomposition of compound 8 was monitored by HPLC. (a) compound 8, (b) compound 8 upon photoirradiation (10 min, 20 mW cm−2), (c) compound 11, (d) compound 3, (e) compound 12. *![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) We could not identify the structure of this product. In all chromatograms, absorption at 254 nm was monitored. (f) Expected photodecomposition mechanism of compound 8 (path I) and putative (path II) photodecomposition mechanism of compound 8 to afford the unexpected oxime compound 12. We could not identify the structure of this product. In all chromatograms, absorption at 254 nm was monitored. (f) Expected photodecomposition mechanism of compound 8 (path I) and putative (path II) photodecomposition mechanism of compound 8 to afford the unexpected oxime compound 12. | ||
Fortunately, this decomposition process does not generate any gaseous or HNO-related small molecules, but instead affords 2-nitrosulfinic acid, a common co-product with the HNO-releasing pathway (Fig. 1f). Although the expected product was a minor one, we next examined HNO release by means of GC-MS analysis of N2O formation via dimerization of HNO. As shown in Fig. 2, the mass chromatogram revealed the formation of N2O from Angeli's salt (AS) (Fig. 2b) or after visible light irradiation (20 mW cm−2, 20 min) of an aqueous solution of compound 8 (Fig. 2d and f). In the absence of photoirradiation, this peak was not observed, indicating that compound 8 is stable and does not release HNO in the absence of photoirradiation under ambient conditions (Fig. 2c). Under the same conditions, compound 9 did not give an apparent N2O peak (Fig. 2e and f). These results indicate that compound 8 releases HNO more efficiently than compound 9, reflecting a previous finding that 2-nitro-N-hydroxybenzensulfonamide has higher HNO-releasing ability than 2-bromo-N-hydroxybenzensulfonamide.8c Thus, the HNO-releasing abilities of compounds 8 and 9 appear to depend on the property of the Piloty's acid derivatives themselves.
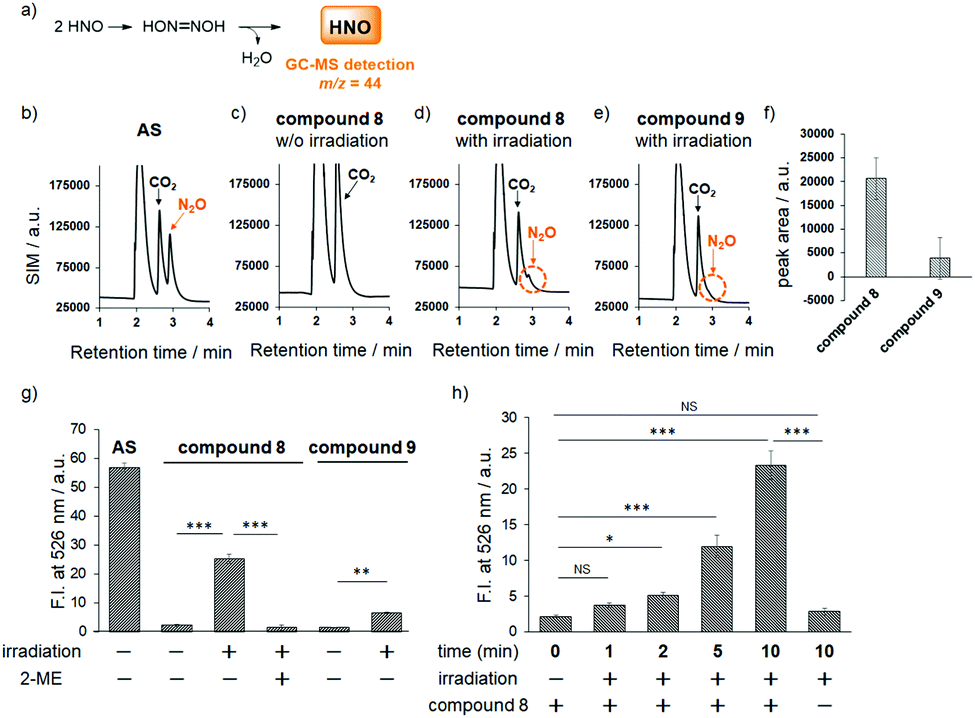 | ||
| Fig. 2 (a) N2O formation from compound 8. GC-MS detection of N2O formation: (b) from AS, (c) from compound 8 in the dark or (d) upon photoirradiation (20 min, 20 mW cm−2), and (e) from compound 9 upon photoirradiation. (f) Integration of the N2O peak (m/z 44). The results are mean ± S.D. (n = 3). (g) Detection of HNO generation using P-Rhod. Compound 8 or 9 (10 μM) was photoirradiated (10 min, 22 mW cm−2) in the presence of P-Rhod. AS (10 μM) was used as a positive control and 2-ME (14 mM) as a HNO scavenger. (h) Irradiation time-dependency of HNO generation. P-Rhod (10 μM) and compound 8 (10 μM) were photoirradiated (22 mW cm−2) for 0, 1, 2, 5, or 10 minutes. The results are mean ± S.D. (n = 3). NS = not significant, *P ≦ 0.05, **P ≦ 0.01, ***P ≦ 0.001 (one-way ANOVA with Bonferroni correction). | ||
Next, to further confirm HNO production from our photocontrollable HNO donors, we used the HNO-specific fluorescence probe, P-Rhod,15 which employs a Staudinger-like reaction for HNO detection. A fluorescence increment was observed only when an aqueous solution containing compound 8 and P-Rhod was photoirradiated (22 mW cm−2 for 10 min; Fig. 2g). In contrast, the fluorescence increment was suppressed in the presence of 2-mercaptoethanol (2-ME), an HNO scavenger. In addition, in the case of compound 9, a smaller fluorescence increment was observed, which is consistent with the GC-MS analyses (Fig. 2a–f). We also examined the irradiation time-dependency of HNO generation and found that compound 8 released HNO in a time-dependent manner (Fig. 2h). From this result, we speculated that the HNO releasing from compound 8 upon photoirradiation is slow enough to be detected with P-Rhod and most of released HNO were detected by P-Rhod.
In addition, the results in Fig. 2 indicate that 10 μM compound 8 released 44% of the amount of HNO released by 10 μM Angeli's salt (AS), meaning that compound 8 would release 4.4 μM HNO, suggesting moderate efficiency. However, this calculation may not be accurate, because AS releases almost all the HNO molecules in water at once, so it is possible that not all the released HNO can react with P-Rhod due to the extremely high reaction rate of HNO dimerization; i.e., a significant amount of dimerization to form N2O may occur, reducing the fluorescence increment with P-Rhod. Therefore, the actual concentration of HNO generated from 10 μM compound 8 is likely to be less than 4.4 μM. Thus, we examined HNO formation from compound 8 upon photoirradiation by means of LC-MS analysis. In this analysis, we detected N,N′-diacetyl-L-cystine formation via reaction between excess N-acetylcysteine (NAC) and HNO, because it is known that thiols react quickly with HNO to afford two products, sulfinamide and disulfide compounds (Fig. S4, ESI†).16 We found that 100 μM compound 8 released an amount of HNO that corresponded to the HNO formation from 16 μM AS. Thus, although the total amount of HNO released from compound 8 was smaller than that from AS, these results confirm that compound 8 can release HNO gradually depending on photoirradiation time (Fig. 2h), which seems consistent with the putative biological actions of HNO. Although endogenous HNO sources have not been elucidated, the reaction of NO and H2S has drawn attention as a potential candidate for HNO formation.17 Because these gaseous molecules, NO and H2S, are enzymatically produced in biological systems, HNO seems to be generated gradually through the chemical reaction between NO and H2S and may work as a signaling molecule. In contrast, a significant amount of HNO generated from AS dimerizes, and thus cannot work as a signaling molecule.
To examine whether compound 8 releases HNO in cellulo upon photoirradiation, we first checked its cell-membrane permeability and subcellular localization. Compound 8 was found to be membrane-permeable, and was mainly localized to endoplasmic reticulum (ER), as judged from the result of co-staining with ER-Tracker Green®; the Manders' overlap coefficient was calculated to be 0.990 (Fig. S5, ESI†). We next treated HEK293T cells (RIKEN Cell Bank, RCB2202) with compound 8 and P-Rhod, and irradiated the cells with visible light (400–430 nm, 20 mW cm−2) for 30 min. Fluorescence intensity was found to be increased in the irradiated cells, as observed by fluorescence microscopy (Fig. 3). In the absence of compound 8, on the other hand, slight fluorescence was observed even after photoirradiation. These results suggest that compound 8 can release HNO in cellulo. Finally, we examined the cytotoxicity and stability of compound 8 in cellular systems and found that compound 8 was almost non-cytotoxic at up to 32 μM (Fig. S6, ESI†) and was completely stable in DMEM containing 5% FBS for at least 24 h in the dark (Fig. S7, ESI†).
 | ||
| Fig. 3 Fluorescence images of HEK293T cells treated with P-Rhod (1 μM) in the presence or absence of compound 8 (20 μM) upon photoirradiation (400–430 nm) for 30 minutes. (a and d) DIC images of cells treated with P-Rhod in the presence or absence of compound 8, respectively. (b and c) Fluorescence images of cells treated with compound 8 and P-Rhod after and before photoirradiation, respectively. (e) Fluorescence image of cells treated with P-Rhod after photoirradiation without compound 8. | ||
In summary, we have designed and synthesized the first nitroxyl (HNO) releasers controllable with visible light (400–430 nm), based on caged Piloty's acid. Although the photodecomposition reaction did not follow the expected path, we demonstrated that the compounds work as photocontrollable HNO releasers and we clarified the actual mechanism of decomposition. Even though HNO is very unstable, we were able to confirm HNO release from compound 8 upon photoirradiation in vitro and in cellulo by means of GC-MS analysis, and by employing an HNO-specific fluorescence probe, P-Rhod. Thus, compound 8 is the first photocontrollable HNO releaser that is applicable in living cells, and we anticipate that it will become indispensable for future biological studies of HNO.
We thank all the members of H. N.'s laboratory. This work was supported in part by JSPS KAKENHI Grant Number 26893223, 15K18899 (M. K.) and 16H05103 (H. N.), as well as by a grant-in-aid for Scientific Research on Innovative Areas from MEXT (26111012, H. N.) and a grant from Daiichi Sankyo Foundation of Life Science (H. N.).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. M. Fukuto, C. H. Switzer, K. M. Miranda and D. A. Wink, Annu. Rev. Pharmacol. Toxicol., 2005, 45, 335–355 CrossRef PubMed; (b) J. C. Irvine, R. H. Ritchie, J. L. Favaloro, K. L. Andrew, R. E. Widdop and B. K. Kemp-Harper, Trends Pharmacol. Sci., 2008, 29, 601–608 CrossRef PubMed; (c) J. M. Fukuto, C. L. Bianco and T. A. Chavez, Free Radical Biol. Med., 2009, 47, 1318–1324 CrossRef PubMed; (d) B. K. Kemp-Harper, Antioxid. Redox Signaling, 2011, 14, 1609–1613 CrossRef PubMed.
- (a) W. D. Gao, C. I. Murray, Y. Tian, X. Zhong, J. F. Dumond, X. Shen, B. A. Stanley, D. B. Foster, D. A. Wink, S. B. King, J. E. Van Eyk and N. Paolocci, Circ. Res., 2012, 111, 1002–1011 CrossRef PubMed; (b) E. Cheong, V. Tumbev, J. Abramson, G. Salama and D. A. Stoyanovsky, Cell Calcium, 2005, 37, 87–96 CrossRef PubMed; (c) C. G. Tocchetti, W. Wang, J. P. Froehlich, S. Huke, M. A. Aon, G. M. Wilson, G. Di Benedetto, B. O'Rourke, W. D. Gao, D. A. Wink, J. P. Toscano, M. Zaccolo, D. M. Bers, H. H. Valdivia, H. Cheng, D. A. Kass and N. Paolocci, Circ. Res., 2007, 100, 96–104 CrossRef PubMed.
- (a) J. A. Norris, M. R. Santippour, M. Lu, T. Park, J. Y. Rao and M. I. Jackson, Int. J. Cancer, 2008, 122, 1905–1910 CrossRef PubMed; (b) N. Paolocci, I. Jackson, B. E. Lopez, K. M. Miranda, C. G. Tocchetti, D. A. Wink, A. J. Hobbs and J. M. Fukuto, Pharmacol. Ther., 2007, 113, 442–458 CrossRef PubMed; (c) C. H. Switzer, W. Flores-Santana, D. Mancardi, S. Donzelli, D. Basudhar, L. A. Ridnour, K. M. Miranda, J. M. Fukuto, N. Paolocci and D. A. Wink, Biochim. Biophys. Acta, Bioenerg., 2009, 1787, 835–840 CrossRef PubMed.
- M. I. Jackson, T. H. Han, L. Serbulea, A. Dutton, E. Ford, K. M. Miranda, K. N. Houk, D. A. Wink and J. M. Fukuto, Free Radical Biol. Med., 2009, 47, 1130–1139 CrossRef PubMed.
- (a) J. A. Reisz, E. Bechtold and S. B. King, Dalton Trans., 2010, 39, 5203–5212 RSC; (b) D. A. Bazylinski and T. C. Hollocher, J. Am. Chem. Soc., 1985, 107, 7982–7986 CrossRef; (c) A. C. Montenegro, V. T. Amorebieta, L. D. Slep, D. F. Martín, F. Roncaroli, D. H. Murgida, S. E. Bari and J. A. Olabe, Angew. Chem., Int. Ed., 2009, 48, 4213–4216 CrossRef PubMed.
- (a) D. A. Bazylinski and T. C. Hollocher, Inorg. Chem., 1985, 24, 4285–4288 CrossRef; (b) V. Shafirovich and S. V. Lymar, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7340–7345 CrossRef PubMed.
- A. Angeli, Gazz. Chim. Ital., 1896, 26, 17 Search PubMed.
- (a) O. Piloty, Ber. Dtsch. Chem. Ges., 1896, 29, 1559 CrossRef; (b) M. R. Cline, C. Tu, D. N. Silverman and J. P. Toscano, Free Radical Biol. Med., 2011, 50, 1274–1279 CrossRef PubMed; (c) K. Izawa, H. Nakagawa, K. Matsuo, K. Kawai, N. Ieda, T. Suzuki and N. Miyata, Bioorg. Med. Chem. Lett., 2013, 23, 2340–2343 CrossRef PubMed.
- (a) K. M. Miranda, T. Katori, C. L. Torres de Holding, L. Thomas, L. A. Rindnour, W. J. McLendon, S. M. Cologna, A. S. Dutton, H. C. Champion, D. Mancardi, C. G. Tocchetti, J. E. Saavedra, L. K. Keefer, K. N. Houk, J. M. Fukuto, D. A. Kass, N. Paolocci and D. A. Wink, J. Med. Chem., 2005, 48, 8220–8228 CrossRef PubMed; (b) L. K. Keefer, ACS Chem. Biol., 2011, 6, 1147–1155 CrossRef PubMed.
- (a) R. N. Atkinson, B. M. Storey and S. B. King, Tetrahedron Lett., 1996, 52, 9287–9290 CrossRef; (b) Y. Xu, M. M. Alavanya, V. L. Johnson, G. Yasaki and S. B. King, Tetrahedron Lett., 2000, 41, 4265–4269 CrossRef; (c) B. B. Zeng, J. Huang, M. W. Wright and S. B. King, Bioorg. Med. Chem. Lett., 2004, 14, 5565–5568 CrossRef PubMed.
- X. Sha, T. S. Isbell, R. P. Patel, C. S. Day and S. B. King, J. Am. Chem. Soc., 2006, 128, 9687–9692 CrossRef PubMed.
- (a) Y. Adachi, H. Nakagawa, K. Matsuo, T. Suzuki and N. Miyata, Chem. Commun., 2008, 5149–5151 RSC; (b) K. Matsuo, H. Nakagawa, Y. Adachi, E. Kameda, H. Tsumoto, T. Suzuki and N. Miyata, Chem. Commun., 2010, 46, 3788–3790 RSC; (c) H. Nakagawa, Nitric oxide, 2011, 25, 195–200 CrossRef PubMed; (d) H. Nakagawa, J. Inorg. Biochem., 2013, 118, 187–190 CrossRef PubMed.
- Y. Zhou, R. B. Cink, R. S. Dassanayake, A. J. Seed, N. E. Brasch and P. Sampson, Angew. Chem., Int. Ed., 2016, 55, 13229–13232 CrossRef PubMed.
- (a) T. Eckardt, V. Hagen, B. Schade, R. Schmidt, C. Schweitzer and J. Bendig, J. Org. Chem., 2002, 67, 703–710 CrossRef PubMed; (b) N. Ieda, S. Yamada, M. Kawaguchi, N. Miyata and H. Nakagawa, Bioorg. Med. Chem., 2016, 24, 2789–2793 CrossRef PubMed.
- K. Kawai, N. Ieda, K. Aizawa, T. Suzuki, N. Miyata and H. Nakagawa, J. Am. Chem. Soc., 2013, 135, 12690–12696 CrossRef PubMed.
- W. D. Gao, C. I. Murray, Y. Tian, X. Zhong, J. F. DuMond, X. Shen, B. A. Stanley, D. B. Foster, D. A. Wink, S. B. King, J. E. Van Eyk and N. Paolocci, Circ. Res., 2012, 111, 1002–1011 CrossRef PubMed.
- Z. Miao and S. B. King, Nitric oxide, 2016, 57, 1–14 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc04954h |
| This journal is © The Royal Society of Chemistry 2018 |