
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An integrated total neutron scattering – NMR approach for the study of heterogeneous catalysis†
Markus
Leutzsch‡
 a,
Marta
Falkowska‡
b,
Terri-Louise
Hughes‡
b,
Andrew J.
Sederman
a,
Lynn F.
Gladden
a,
Michael D.
Mantle
*a,
Tristan G. A.
Youngs
c,
Daniel
Bowron
c,
Haresh
Manyar
d and
Christopher
Hardacre
*b
a,
Marta
Falkowska‡
b,
Terri-Louise
Hughes‡
b,
Andrew J.
Sederman
a,
Lynn F.
Gladden
a,
Michael D.
Mantle
*a,
Tristan G. A.
Youngs
c,
Daniel
Bowron
c,
Haresh
Manyar
d and
Christopher
Hardacre
*b
aDepartment of Chemical Engineering and Biotechnology, Philippa Fawcett Drive, University of Cambridge, Cambridge, CB3 0AS, UK. E-mail: mdm20@cam.ac.uk
bSchool of Chemical Engineering and Analytical Science, The University of Manchester, Manchester, M13 9PL, UK. E-mail: c.hardacre@manchester.ac.uk
cSTFC ISIS Facility, Rutherford Appleton Laboratory Chilton, Didcot, Oxon OX1 10QX, UK
dSchool of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast BT9 5AG, UK
First published on 23rd August 2018
Abstract
Nuclear magnetic resonance (NMR) and total neutron scattering techniques are established methods for the characterisation of liquid phases in confined pore spaces during chemical reactions. Herein, we describe the first combined total neutron scattering – NMR setup as a probe for the catalytic heterogeneous reduction of benzene-d6 with D2 in 3 wt% Pt/MCM-41.
Catalytic processes play significant roles in the pharmaceutical, bulk and fine chemical industries as well as in food processing and emission control. In the majority of these applications heterogeneous catalysts are used.1 Despite their frequent use, a full understanding of the underlying microscopic and macroscopic processes is limited due to the complex nature of these reactions. Besides the reactivity of the active catalytic centre, other processes including adsorption and desorption, interactions of reactants and products with the surface or mass transport to the active centre are often rate limiting steps of these systems. Over the past decades, multidisciplinary research has helped to understand individual components of heterogeneous processes. Analytical diffraction and spectroscopy techniques have been used to examine in detail the properties of the catalyst material or processes that occur during heterogeneously catalysed reactions.2,3 Total neutron scattering has recently been shown to be able to probe the arrangement of liquid reagent molecules within the pores of a catalyst support.4 This technique can also be used to monitor changes on a molecular and nanometric length scale during chemical reactions within catalyst pores, and thus to give insight into kinetics of heterogeneously catalysed processes.5,6 The kinetic information can be obtained directly from experimentally measured neutron scattering data without any numerical modelling. However, in order to investigate the interactions between molecules, detailed modelling of the data is required. Empirical Potential Structure Refinement (EPSR)7,8 enables the generation of a three-dimensional model of a system of interest and optimizes it by comparing the generated structures to the experimental neutron diffraction data. Modelling of reacting mixtures requires detailed compositional information at a given point in time, which cannot be obtained from total neutron scattering or ex situ sampling methods due to sensitivity, radiation and experimental setup restrictions. Recently, in situ NMR techniques have not only delivered insights into spatial and chemical resolved changes in heterogeneous reactors at operando conditions,9,10 but have also been demonstrated to be able to examine solvent effects,11 explain reactivity trends and adsorption strength via T1 and T2 relaxation12 and measure the diffusion of reactants and products in porous materials.10 In most of the examples, these investigations deliver only a small piece of an often more complicated puzzle. The interpretation of data for heterogeneous systems from individual analytical techniques, for nominally the same reaction, can be challenging as one usually has to examine the system under slightly different conditions due to the different experimental setups. Thus it can be advantageous to incorporate modalities in one experimental setup. For example, NMR has recently been combined with other spectroscopic techniques, such as UV-Vis-13 or Raman-Spectroscopy14 in a single apparatus.
Inspired by these advances, this paper describes the development of a coupled total neutron scattering – NMR (NeuNMR) setup that enables a better understanding of the liquid phase of heterogeneously catalysed systems by combining the information from both techniques.
Previous research on the hydrogenation of aromatic systems using total neutron scattering revealed different rates of physicochemical processes involving liquid and D2 rearrangements within the pore structure of the supported metal catalyst as well as changes at a molecular level.5,6 However, it is important to note that the information contained within the neutron scattering data relating to chemical changes, mass transport and surface adsorption is frequently non-trivial to extract, as the signals of interest make overlapping contributions to the measured structure factors. Furthermore the technique is limited in its temporal resolution by the radiation fluxes that can be delivered to the samples from the source. With the widespread availability of inexpensive, portable, medium-field high resolution NMR hardware15,16 a natural choice is to merge the structural information available from neutron scattering with the aforementioned quantitative information available from NMR. A schematic drawing of a combined total neutron scattering – NMR setup, that was implemented at the Near and InterMediate Range Order Diffractometer (NIMROD)17 at the ISIS Pulsed Neutron and Muon Source is shown in Fig. 1. Due to the requirements of the neutron scattering data acquisition, the NeuNMR setup had to be contained within a vacuum (0.1 mbar) chamber. As a consequence, some changes had to be made to the commercial NMR System (see ESI† for further information). During the experiment, two concurrently prepared samples from the same batch of catalyst are placed into two separate sampling regions; these are directly connected and are, therefore, under the same environment in terms of pressure and temperature. The neutron measurements are conducted in a 6 mm o.d./5.7 mm i.d. vanadium tube, which has a low scattering background. In contrast to the previous experiments,5,6 the sample container has been changed from a flat plate cell to a cylindrical cell in order to mimic the sample geometry of the NMR sample, which uses a 5 mm thin wall NMR tube.
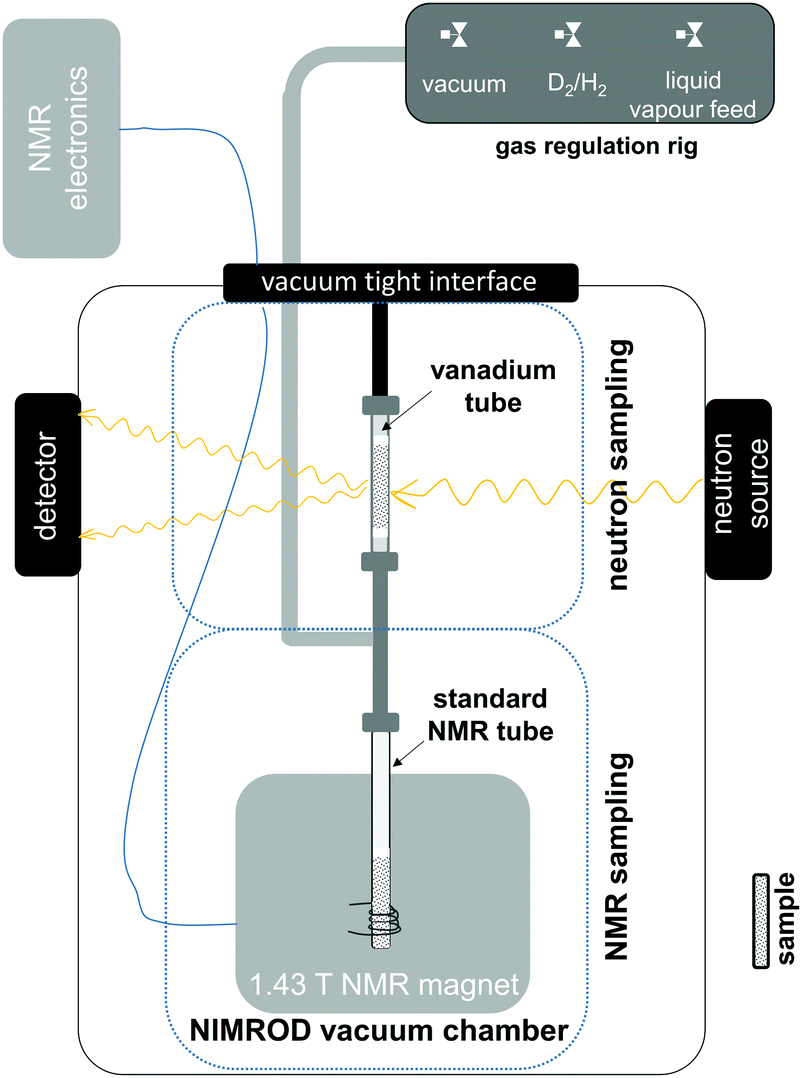 | ||
| Fig. 1 Schematic drawing of the combined total neutron scattering – NMR setup in the Near and Intermediate Range Order Diffractometer at the ISIS Pulsed Neutron and Muon Source. | ||
The hydrogenation of benzene in mesoporous Pt/MCM-41 has been shown previously to be a good benchmark system for following reaction kinetics by total neutron scattering.5 This is due to the high contrast between the local intermolecular ordering of molecules in the final product (cyclohexane-d12) and starting reagent (benzene-d6) as well as intramolecular differences between those two compounds.8 Therefore, this reaction was chosen to test the NeuNMR setup. To obtain the optimal signal-to-noise ratio from the neutron measurements and to prevent H/D-exchange processes, benzene-d6 and D2 instead of their protiated analogues were used as reagents. The initial deposition of benzene-d6 from the gas phase via capillary condensation into the pores was monitored by 2H NMR and total neutron scattering; this pore-filling process and was completed in approx. 15 min (see ESI†). Due to this procedure, the bulk signal detected in both NMR and neutron measurements arises solely from liquid residing within the pore space of the catalyst. Fig. 2a shows the changes in the 2H NMR spectrum at different time points during the hydrogenation of benzene-d6 under an atmosphere of 250 mbar D2 in 3 wt% Pt/MCM-41 at 21.5 °C. The NMR signals of liquids confined in mesoporous materials are, in general, broader than NMR signals in pure liquids due to differences in magnetic susceptibility of the different material phases present. Dynamic exchange between the bulk liquid phase and the solid surface of the porous catalyst reduces the T1 and T2 relaxation rates of the liquid reactants and further depends on the pore size distribution and the strength of interaction between the surface and the molecule.18,19 After an initial reduction period, which led to a slight loss of benzene (δ2H = 7.3 ppm) due to sample heating, the formation of cyclohexane-d12 (δ2H ∼ 1.0 ppm), is observed over a time of 17 h. The 2H NMR signal of benzene-d6 is broader compared to the signal of cyclohexane-d12 for two reasons: (i) the enhanced OD–π interaction with the aromatic ring and the Si–OH groups enhances the average residence time on the surface; (ii) C6D6 has a faster quadrupolar relaxation rate than C6D12. This process is more effective than the dipolar mechanism in the protiated analogue and therefore is easier to observe and results in a broader NMR linewidth. The potential reaction intermediates cyclohexene and cyclohexadiene were not observed during the reaction. The region of total structure factors presented in Fig. 2b represent correlations between nanoscale structural features in the sample, i.e. pore–pore correlations expressed by three Bragg peaks: (100), (110) and (200). The intensity of these peaks is related to the scattering contrast between the pore content and its walls. When pores are filled with benzene-d6, the scattering contrast between the initial reagent and silica walls is ΔSLD = 1.93 × 10−6 Å−2, whereas when they are filled with the final product (cyclohexane-d12) the contrast increases to ΔSLD = 3.24 × 10−6 Å−2. This is indicated by a higher intensity of Bragg peaks from the catalyst filled with cyclohexane-d12 than when it is filled with benzene-d6, thereby an increase of these features is observed during the course of the reaction. Intermolecular correlations in the sample are represented by total structure factor features shown in Fig. 2c. Strong π–π interactions cause benzene molecules to approach closer to one another than cyclohexane molecules do,8 which can be seen as a shift of the peak from 1.35 Å−1 corresponding to benzene–benzene correlation to 1.23 Å−1 for cyclohexane–cyclohexane correlation during the course of reaction (note that as the total structure factor is a function of inverse distance, higher values in Q-space represent shorter separations between molecules). The Fourier transform of the total structure factors is presented in Fig. 2d, and reveals the changing intramolecular correlations in the measured sample during the hydrogenation reaction. An increase of a peak at 1.05 Å during the course of reaction corresponds to an increased number of C–D bonds in the sample, and thereby confirms the product formation. This is also manifested by a disappearance of the peak corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond at 1.46 Å and simultaneous increase of peak corresponding to C–C bond.
C bond at 1.46 Å and simultaneous increase of peak corresponding to C–C bond.
 | ||
| Fig. 2 The hydrogenation of benzene-d6 by D2 investigated by an integrated total neutron scattering – NMR approach: (a) 2H NMR spectra taken at different time points during the reaction; (b) and (c) structure factors measured at different time points during the reaction; the colour change from yellow to black as presented in the colour bar indicates the peak evolution as time increases (d) direct Fourier transform of data shown in (c) and (d); (e) time dependent change of integral extracted from 2H NMR spectra, the (100) Bragg peak intensity the C–D bond peak intensity and the molecular peak intensity (Q = 1.23 Å−1) are obtained total neutron scattering data, as indicated in (b–d); (f) initial rates extracted from (e). | ||
The increase of the 2H NMR signal is dominated by the conversion of starting material, whereas the absolute changes of signal intensity in the total structure factors obtained from total neutron scattering data are not as simple to interpret. In order to compare the kinetic data obtained from the individual methods, the measured NMR integral and the total neutron scattering peak intensities were normalised. Therefore, the integral of the first NMR spectrum was set to 0% and the signal when no further conversion was observed to 100%. The total neutron scattering data were normalised to the same values at identical time points. Measurements of pressure and temperature changes during the reaction support the assumption that the reaction was completed after approx. 16 h (see ESI† for further information). The normalised signal evolution for both datasets is shown in Fig. 2e. The evolution of the 2H NMR integral, the (100) Bragg peak intensity and the molecular peak at 1.23 Å−1 show very similar trends. Data obtained from the C–D bond peak intensity which is directly connected to the product formation is very noisy, but the data is consistent with the other datasets shown. Previous kinetic studies on benzene hydrogenation reactions showed a dependence of the partial hydrogen pressure on the reaction rate.20 In our experiment, a drop of deuterium pressure and therefore a decrease of reaction rate was also observed at higher conversion (see ESI†). Since the mass transport of benzene becomes increasingly rate limiting as the reaction proceeds, to compare the kinetic results from the different techniques, we therefore determined initial rates of the signal evolution as this is not rate limiting in starting material availability.21 The results are presented in Fig. 2f. The evolution of the Bragg peak is expressed by a rate constant of 0.094 h−1. The previous study6 showed that changes in the Bragg peak regions can be assigned to the processes associated with D2 sorption as well as confined liquid reorganisation, including diffusion. The former process was described by a rate constant of 2.146 h−1 and the latter by a rate constant of 0.136 h−1. In the current study, these processes were slower. Furthermore, only one linear rate constant, instead of two exponentials, was required to characterise the reaction during the initial time period. This suggests that the two processes identified have become too similar in terms of their rate constant to be able to differentiate them. A similar issue was observed for toluene hydrogenation studied by total neutron scattering when changing the D2 pressure from 150 mbar to 250 mbar.7 In the data presented here, the cyclohexane formation obtained from the NMR data occurs at an initial rate of 0.083 h−1, whereas changes in the intensity of molecular peak in neutron data are described by a rate of 0.083 h−1. Both rates have very similar values, and this shows the excellent agreement between the NMR and neutron data. The C–D bond evolution, obtained from direct Fourier transform of total structure factor, grows with a rate of 0.096 h−1 and is within the error range of the other rates. The rate constant for benzene hydrogenation obtained previously,5 namely 0.35 h−1 indicates that the formation of cyclohexane in the current study was almost 4 times slower due to different metal loadings and a catalyst preparation procedure.
In summary, this communication describes the first combined total neutron scattering – NMR setup to investigate a liquid phase heterogeneous hydrogenation reaction. The hydrogenation of benzene over 3 wt% Pt/MCM-41 was used as the benchmark reaction and resulted in complementary kinetic data from both techniques. The obtained information can be used as input variables for EPSR simulations of the structuring in the liquid phase at different points of the reaction. This will enable a better understanding of structural changes during these processes and can help to understand the relationship between structural changes in confined environments and reactivity. These results suggest that the NeuNMR methods can be applied to measure more complex multi-pathway reactions to better understand the influence of confinement, selectivity and reactivity, which can help to develop new catalyst materials with better selectivity. The use of NMR to probe these more complex reactions is crucial, as otherwise it is not possible to assign the structural changes observed by neutron scattering to formation of intermediate products. These have often very similar structure and may be indistinguishable in the neutron dataset on its own. Further advances of the technique will include carrying out reactions in continuous flow, which is of great importance in the industrial research and development. 2H NMR can limit the information obtained from this new technique as the chemical shift dispersion is smaller compared to 1H. When H/D exchange processes are not a problem, the use of partially protiated analogues could help overcome these issues in the future and could also enable the use pulsed field gradient NMR techniques to study mass transport changes during the reaction.
The authors would like to thank Science and Technology Facilities Council for the allocated beam time at the ISIS Pulsed Neutron and Muon Source (RB1710475) as well as the funding from EPSRC (EP/N008995/1 and EP/N009304/1, Understanding Liquid Phase Heterogeneous Catalysis to Develop Catalytic Processes). Raw experimental total neutron scattering and NMR data may be accessed at https://doi.org/10.5286/ISIS.E.87813090 and https://doi.org/10.17863/CAM.24725 respectively.
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Handbook of Heterogeneous Catalysis, Wiley-VCH, Weinheim, 2008 Search PubMed.
- W. Delgass, Spectroscopy in Heterogeneous Catalysis, Academic Press, New York, 1979 Search PubMed.
- M. O. Jones, A. D. Taylor and S. F. Parker, Appl. Petrochem. Res., 2012, 2, 97 CrossRef.
- M. Falkowska, D. T. Bowron, H. Manyar, T. G. A. Youngs and C. Hardacre, Angew. Chem., Int. Ed., 2018, 57, 4565 CrossRef PubMed.
- T. G. A. Youngs, H. Manyar, D. T. Bowron, L. F. Gladden and C. Hardacre, Chem. Sci., 2013, 4, 3484 RSC.
- M. Falkowska, S. Chansai, H. G. Manyar, L. F. Gladden, D. T. Bowron, T. G. A. Youngs and C. Hardacre, Phys. Chem. Chem. Phys., 2016, 18, 17237 RSC.
- A. K. Soper, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 104204 CrossRef.
- M. Falkowska, D. T. Bowron, H. G. Manyar, C. Hardacre and T. G. A. Youngs, ChemPhysChem, 2016, 17, 2043 CrossRef PubMed.
- A. J. Sederman, M. D. Mantle, C. P. Dunckley, Z. Huang and L. F. Gladden, Catal. Lett., 2005, 103, 1 CrossRef.
- L. Baker, M. P. Renshaw, M. D. Mantle, A. J. Sederman, A. J. Wain and L. F. Gladden, Appl. Catal., A, 2018, 557, 125 CrossRef.
- C. D’Agostino, T. Kotionova, J. Mitchell, P. J. Miedziak, D. W. Knight, S. H. Taylor, G. J. Hutchings, L. F. Gladden and M. D. Mantle, Chem. – Eur. J., 2013, 19, 11725 CrossRef PubMed.
- Y. Wu, C. D’Agostino, D. J. Holland and L. F. Gladden, Chem. Commun., 2014, 50, 14137 RSC.
- M. Hunger and W. Wang, Chem. Commun., 2004, 584 RSC.
- J. C. J. Camp, M. D. Mantle, A. P. E. York and J. McGregor, Rev. Sci. Instrum., 2014, 85, 063111 CrossRef PubMed.
- E. Danieli, J. Perlo, B. Blümich and F. Casanova, Angew. Chem., Int. Ed., 2010, 49, 4133 CrossRef PubMed.
- B. Blümich and K. Singh, Angew. Chem., Int. Ed., 2018, 57, 6996 CrossRef PubMed.
- D. T. Bowron, A. K. Soper, K. Jones, S. Ansell, S. Birch, J. Norris, L. Perrott, D. Riedel, N. J. Rhodes, S. R. Wakefield, A. Botti, M.-A. Ricci, F. Grazzi and M. Zoppi, Rev. Sci. Instrum., 2010, 81, 033905 CrossRef PubMed.
- D. Weber, J. Mitchell, J. McGregor and L. F. Gladden, J. Phys. Chem. C, 2009, 113, 6610 CrossRef.
- C. D’Agostino, J. Mitchell, M. D. Mantle and L. F. Gladden, Chem. – Eur. J., 2014, 20, 13009 CrossRef PubMed.
- U. K. Singh and M. A. Vannice, AIChE J., 1999, 45, 1059 CrossRef.
- J. Casado, M. A. Lopez-Quintela and F. M. Lorenzo-Barral, J. Chem. Educ., 1986, 63, 450 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc04740e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |