
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn-surface synthesis of heptacene on Ag(001) from brominated and non-brominated tetrahydroheptacene precursors†
Luciano
Colazzo
ab,
Mohammed S. G.
Mohammed
ab,
Ruth
Dorel
 c,
Pawel
Nita
ab,
Carlos
García Fernández
a,
Paula
Abufager
d,
Nicolás
Lorente
ab,
Antonio M.
Echavarren
ce and
Dimas G.
de Oteyza
*abf
c,
Pawel
Nita
ab,
Carlos
García Fernández
a,
Paula
Abufager
d,
Nicolás
Lorente
ab,
Antonio M.
Echavarren
ce and
Dimas G.
de Oteyza
*abf
aDonostia International Physics Center (DIPC), 20018 San Sebastián, Spain. E-mail: d_g_oteyza@ehu.es
bCentro de Física de Materiales (CSIC-UPV/EHU) – MPC, 20018 San Sebastián, Spain
cInstitute of Chemical Research of Catalonia (ICIQ), Barcelona Institute of Science and Technology, 43007 Tarragona, Spain
dInstituto de Física de Rosario, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) and Universidad Nacional de Rosario, (2000) Rosario, Argentina
eDepartament de Química Orgànica i Analítica, Universitat Rovira i Virgili, 43007 Tarragona, Spain
fIkerbasque, Basque Foundation for Science, 48011 Bilbao, Spain
First published on 28th August 2018
Abstract
Achieving the Ag(001)-supported synthesis of heptacene from two related reactants reveals the effect of the presence of Br atoms on the reaction process. The properties of reactants, intermediates and end-products are further characterized by scanning tunneling microscopy and spectroscopy.
Acenes are a class of polycyclic aromatic hydrocarbons consisting of linearly fused benzene rings. Their interesting electronic properties have made them the subject of a vast number of experimental and theoretical studies.1 For example, pentacene has been integrated into several types of devices with remarkable success because of its particularly high charge carrier mobility.1 Acenes longer than pentacene are predicted to have even more promising properties,2 but have been studied much less. This is directly related to their lower stability,3 which makes their synthesis by conventional wet chemistry much more challenging,4 and further limits their potential implementation into device structures. Indeed, the synthesis of non-substituted higher acenes has been until recently restricted to the photodecarbonylation of carbonyl-bridged precursors, and needs to be carried out in stabilizing inert matrices at cryogenic temperatures.5 Among the reasons for such reduced stability is the increasing open-shell character predicted for acenes as they grow longer. According to calculations, partial diradical character starts appearing for hexacene, with a notable contribution of polyradical character for longer acenes like undecacene or dodecacene.6 That is, predictions associate every five to six rings with roughly two unpaired electrons.7
On-surface chemistry under ultra-high vacuum (UHV) has appeared as an efficient way to overcome some of these limitations. Under the clean and controlled environment of UHV chambers, higher acenes have not only been successfully synthesized, but are also stable, allowing their subsequent characterization with surface science techniques. Following this approach, tetracene,8 hexacene,9 heptacene,10,11 octacene,11 nonacene,11,12 decacene11,13 and undecacene11 have been synthesized and characterized on Au(111). Pentacene and heptacene have also been synthesized on Ni(111)14 and Ag(111),15 respectively.
In this work, starting from two different precursors, we study the synthesis process of heptacene on Ag(001). Single molecule analysis by scanning tunneling microscopy (STM) and scanning tunneling spectroscopy (STS) has been applied to extract information on the structural and electronic properties of reactants, intermediates, and heptacene end-products.
The precursors correspond to dibromotetrahydroheptacene (Br-1, Fig. 1a) and tetrahydroheptacene (1, Fig. 1d),16 merely differing in the bromination (or its absence) at the central carbon ring. Deposition of the reactants on a surface held at room temperature (RT) and subsequently cooled to 4.3 K for their characterization results in samples as shown in Fig. 1. The samples with Br-1 (Fig. 1b and c) display two types of well-differentiated surface morphologies: reconstructed substrate regions with highly periodic monatomic steps along the compact [110] or [1−10] directions (see also Fig. S1, ESI†) and flat Ag(001) terraces. Unless high coverages force the formation of close-packed layers all over the substrate, the molecules appear preferentially adsorbed on the reconstructed regions. All the molecules appear readily debrominated at RT. While this is similar to the findings with other brominated reactants on Ag(110),17 it differs from the results on Ag(111),17,18 suggesting, as generally expected, a higher reactivity of less densely packed surfaces. Each reconstructed terrace fits one molecule and one to three neighbouring Br atoms. The molecules on subsequent terraces appear aligned with respect to one another and display long-range order domains.
 | ||
| Fig. 1 (a) Chemical structure of precursor Br-1 and representative small (b) and large (c) scale STM images of the surface after its deposition on Ag(001) held at RT [U = 30 mV/I = 70 pA (b) and U = 30 mV/I = 100 pA (c)]. (d) Chemical structure of reactant 1 and representative small (e) and large (f) scale STM images of the sample after its deposition on Ag(001) [U = 35 mV/I = 100 pA (e) and U = 600 mV/I = 600 pA (f)]. | ||
Equivalent experiments with the non-halogenated reactant 1 lead to notably different results (Fig. 1d and e). In the absence of Br substituents, the molecules on the Ag(001) terraces align along the [110] or [1−10] directions, but cause neither a surface reconstruction nor show any kind of long-range order. We thus associate these two effects to Br-driven interactions, in line with previous reports of other brominated precursors on noble metal surfaces.19
Upon heating, both kinds of reactants suffer dehydrogenation of the non-aromatic rings. In this process, hydrogens are lost pairwise, while on the organic scaffold the aromatization of the rings occurs as displayed in Fig. 2. Focusing first on Br-1, a stepwise dehydrogenation is observed for the two rings. Although a detailed characterization of threshold temperatures was not performed, annealing of the samples to 180 °C caused the aromatization of one ring in most of the molecules (Fig. 3a), and a quasi-full transformation into heptacene was observed after annealing to 270 °C. The disparate activation temperatures evidence differences in the mechanisms of the first and second aromatization events, which may be affected e.g. by the potential remaining radicals generated upon dehalogenation. After aromatization of the first ring, the remaining non-aromatic ring easily shifts along the molecule (Fig. 2h–i). Its position can be distinguished through high-resolution constant height imaging using CO-functionalized tips, which features methylene functionalities on polyacenes as distinctly wider rings (Fig. 2a and b). Alternatively, an indentation-like contrast at low bias (highlighted with arrows in Fig. 2d–i), an increased apparent height at positive bias or a node at negative bias (Fig. S2, ESI†) is observed at the hydrogenated rings by conventional constant current STM imaging. A statistical analysis of the position of the hydrogenated ring reveals its tendency to shift toward the central part of the molecule (Fig. 3a and b). Since the hydrogenation breaks the conjugation along the molecule, intermediates 2–4 (Fig. 2) can be seen as made up by two coupled but independent acenes. Taking into account that the aromatic stabilization energy per π-electron as calculated from homodesmic reactions is notably reduced as the number of annulated rings increases,20 the shorter the acene segments to be combined are, the more stable the molecules are. As a result, their occurrence is the highest for 4, followed by 3 and then by 2, whose longest acene segments correspond to anthracene, tetracene and pentacene, respectively. In fact, even those few molecules that retain tetrahydrogenation after this first annealing step (e.g. oval in Fig. 3a) display the hydrogenation preferentially at rings 2 and 3 on either side of the molecular center (intermediate 1′, Fig. 2g), reducing the maximum conjugation length from an anthracene to a naphthalene segment. If heated to higher temperatures (e.g. 270 °C), the remaining non-aromatic ring also gets dehydrogenated, rendering heptacene as end-product 5 (Fig. 2c and f).
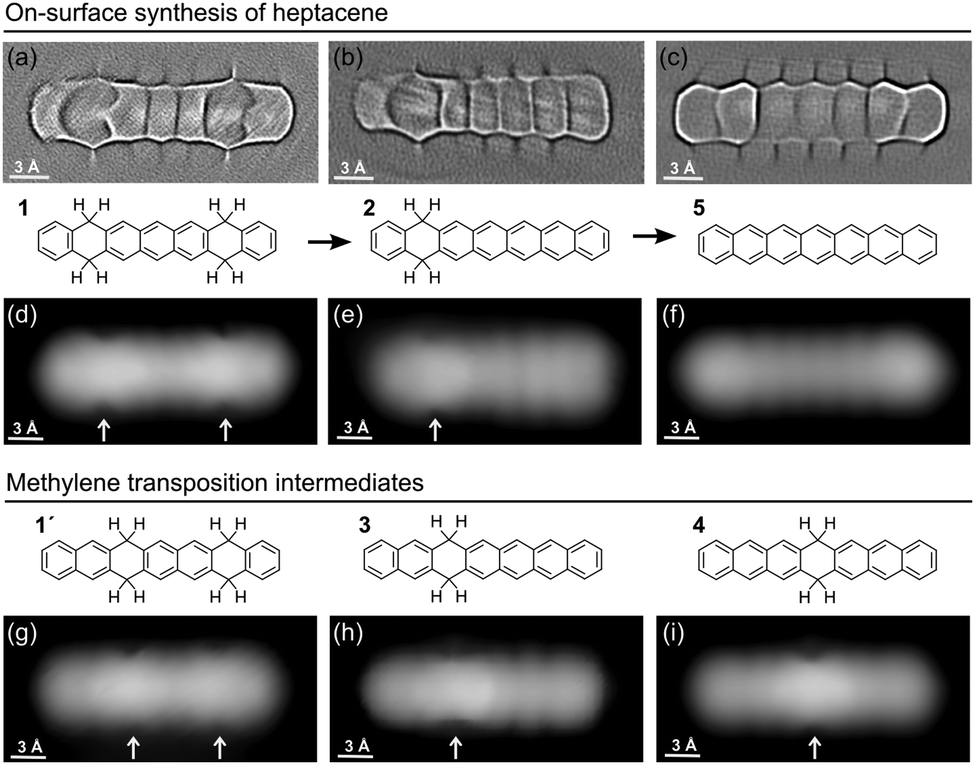 | ||
| Fig. 2 On-surface synthesis process of heptacene from Br-1, displaying the evolution of the molecule's chemical structure throughout the reaction with two, one, and no hydrogenated non-aromatic rings. The associated high resolution and constant height images revealing the bonding structures (U = 2 mV, Laplace filtered) are shown in panels (a), (b) and (c), respectively. Panels (d), (e) and (f) display the corresponding constant current STM images (U = 30–50 mV/I = 100 pA), while (g), (h) and (i) display the intermediates involving methylene transposition. All images are recorded with CO-functionalized probes. | ||
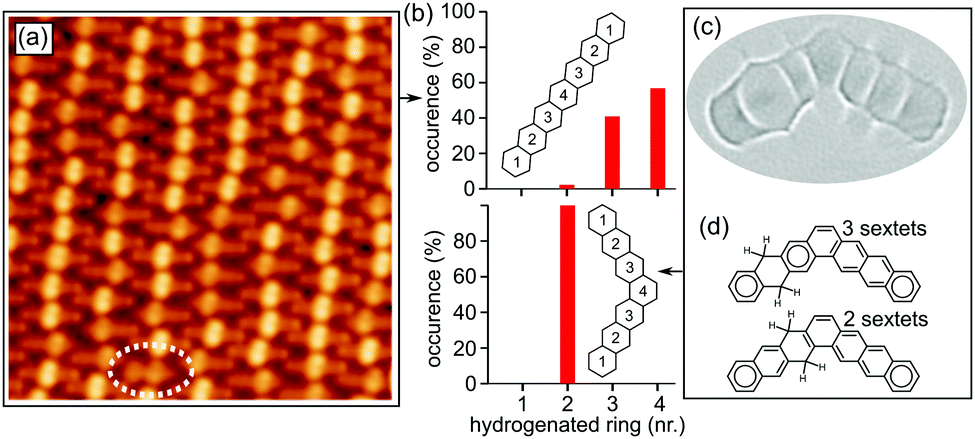 | ||
| Fig. 3 (a) STM image (U = 1.7 V/I = 186 pA) of a full monolayer sample of Br-1 after annealing to 180 °C. The oval marks a tetrahydro-heptacene molecule after a methylene migration (intermediate 1′). (b) Histograms displaying the occurrence of differently positioned hydrogenated rings (numbered as in the insets of molecular structures) for dihydro intermediates of heptacene (top) and heptaphene (bottom). (c) Laplace-filtered constant-height STM image (U = 2 mV) of a heptaphene intermediate with a single hydrogenated ring. (d) Schematics revealing the maximized number of Clar sextets for dihydroheptaphene when hydrogenated at the second ring (compared below, e.g., with hydrogenation at the third ring). | ||
Interestingly, the scenario changes substantially in the case of “kinked acene isomers” (Fig. 3c). Analyzing a sample containing minor amounts of tetrahydroheptaphene (see the ESI† for details), we observed that, in contrast to the linear counterparts, the saturation of the dihydro intermediates always remains at ring 2 (Fig. 3b). In this case, any hydrogen migration would reduce the number of Clar sextets in the molecule from three to two (Fig. 3d), corroborating that the stabilization energy per Clar sextet20,21 makes the configuration with the methylenes on ring 2 most favorable.
Also the non-halogenated precursor 1, still following a similar reaction process as Br-1, displays notable differences. As opposed to the Br-1 case displayed in Fig. 3a, a similar annealing to 180 °C results in an approximately 80/13/7 ratio of the tetrahydro, dihydro and heptacene species (Fig. S3, ESI†). That is, at the same temperature, less molecules display any aromatization, and out of those that do, more aromatize fully into 5. Besides, the formal methylene migration on reactants and dihydro-intermediates is much less common when starting from 1. Thus, although the same tendency to minimize the conjugated acene segments through formal methylene migration was reported for closely related non-brominated tetrahydrononacene molecules on Au(111),12 and although an analysis of the detailed mechanism involved in these chemical changes is beyond the scope of this paper, it seems evident that the initial presence of radicals after dehalogenation facilitates the first aromatization and lowers the hydrogen transposition barriers.
Within these experiments, we end up with a plethora of different but closely related molecules, offering a good opportunity to probe structure–property relations. At this stage it is important to note that, although with slight differences in the reaction process described above, the structural and electronic characterization performed on single molecules on Ag(001) terraces is similar regardless of whether 1 or Br-1 is used. That is, we observe the products from a spontaneous hydrogenation of the radical sites resulting from the dehalogenation of Br-1. This is of key importance in the analysis and understanding of many surface-supported reactions under vacuum and may be rationalized taking into consideration that hydrogen makes up for most of the residual gas present in UHV chambers and may be additionally generated by nearby dehydrogenation events.
We have recorded dI/dV STS spectra on each of those different molecules for comparison. The results are summarized in Fig. 4a, placed in order according to the molecule's longest conjugated segment, which is known to host most of the electron densities of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbitals (LUMO) (Fig. S4, ESI†).12 Thus, we start from 1′, displaying naphthalene as the largest conjugated segment, followed by 1 and 4 (anthracene), 3 (tetracene), 2 (pentacene) and 5 (heptacene). For the sake of comparison, we also add heptaphene (6), the fully conjugated kinked isomer of heptacene (5).
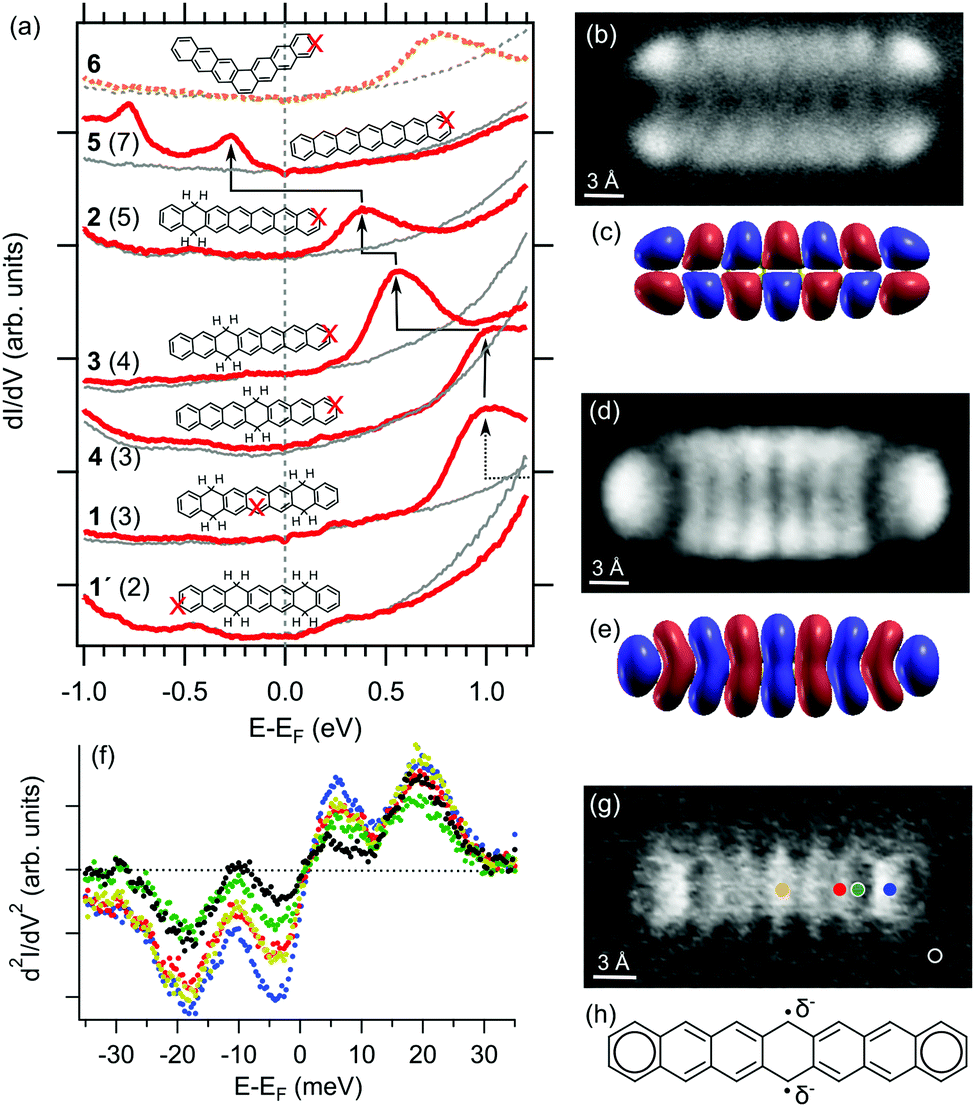 | ||
| Fig. 4 (a) dI/dV point spectra on reactants, intermediates and end-products, as well as heptaphene 6 (colored lines) at positions as marked on the accompanying models and the associated reference substrate spectra (grey lines). The number of rings in the longest conjugated segment is given in brackets and the LUMO position is marked with arrows. Conductance maps (I = 180 pA, Uosc = 10 mV, and ν = 341 Hz) of 5 at −0.76 V (b) as well as at −0.25 V (d) and its calculated gas-phase wavefunction of the HOMO (c) and the LUMO (e). (f) d2I/dV2 spectra (Uosc = 2 mV and ν = 341 Hz) at the positions marked in (g), revealing the frustrated translational and rotational modes of the CO on the functionalized tip at around 3 and 20 meV, respectively. (g) Inelastic electron tunneling spectroscopy map (const. z, Uosc = 7 mV, and ν = 341 Hz) of 5 at 3.5 mV. (h) Schematic model of the open-shell heptacene structure, with the associated radicals partially stabilized by additional electron charges. | ||
The HOMO–LUMO gap is known to decrease with the size of acenes,5c,11,12 as also reproduced with density functional theory calculations of these specific molecules (Fig. S4, ESI†). Although experimentally we have only accessed the LUMO for most of the probed molecules, it can be nicely observed how it approaches the Fermi level as the largest conjugated segment increases in size from 1 to 2.22,23 Following the same trend, the shift from 2 to 5 occurs by crossing the Fermi level, concurrently with the appearance of a second occupied resonance. Conductance maps at the corresponding energies (Fig. 4b and d) reveal their resemblance with the calculated gas-phase wavefunctions of the HOMO and LUMO (Fig. 4c and e), with slight differences in representations stemming from the mixed s- and p-wave character of the CO-functionalized probe used for the characterization.24 These data are thus clear evidence of the charging of heptacene on Ag(001) by the filling of its former gas-phase LUMO.
Such important charge transfer suggests a strong molecule–substrate interaction,25 as opposed to weakly interacting interfaces that rather display a Fermi-level pinning scenario.26 The strong interaction is proposed to be promoted by the partial open-shell character of heptacene, whose radicals could be stabilized by the additional electrons as schematically shown in Fig. 4h. Experimental findings hinting at this particular scenario are the spatial variations in the vibrations of CO at functionalized tips when performing inelastic electron tunneling spectroscopy (IETS).27 Such vibrations, and in particular the frustrated translational mode occurring at around 3 mV at the silver–CO–tip gap, are strongly dependent on the charge density of the probed sample and show the strongest intensity variations at the outer and central rings (Fig 4f and g), where the Clar sextets and the partial radical character are expected to be dominantly located.
Altogether, we have studied the surface-supported synthesis of heptacene on Ag(001). The use of two different precursors has allowed us to learn about the effect of bromine atoms on the properties of molecules and substrates, as well as about the spontaneous hydrogenation of radical sites even under UHV. Spectroscopic analysis of the reactants, intermediates and end-products has evidenced a change in the energy-level alignment associated with the decreasing band gap of the molecules with increasing conjugation size. This trend ends with a charged heptacene, indicative of a strong interaction with the underlying Ag(001) substrate.
We acknowledge funding from the European Research Council under the European Union's Horizon 2020 programme (Grant Agreement No. 635919), and from the Spanish Ministry of Economy, Industry and Competitiveness (MINECO, Grant No. MAT2016-78293-C6, MAT2015-66888-C3-2-R), Agencia Estatal de Investigación (AEI)/FEDER, UE (CTQ2016-75960-P), the AGAUR (2017 SGR 1257), and the CERCA Program/Generalitat de Catalunya.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef PubMed; (b) M. Bendikov, F. Wudl and D. F. Perepichka, Chem. Rev., 2004, 104, 4891–4946 CrossRef PubMed.
- J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452–483 CrossRef PubMed.
- (a) S. S. Zade and M. Bendikov, J. Phys. Org. Chem., 2012, 25, 452–461 CrossRef; (b) S. S. Zade, N. Zamoshchik, A. R. Reddy, G. Fridman-Marueli, D. Sheberla and M. Bendikov, J. Am. Chem. Soc., 2011, 133, 10803–10816 CrossRef PubMed.
- R. Dorel and A. M. Echavarren, Eur. J. Org. Chem., 2017, 14–24 CrossRef PubMed.
- (a) R. Mondal, B. K. Shah and D. C. Neckers, J. Am. Chem. Soc., 2006, 128, 9612–9613 CrossRef PubMed; (b) C. Tönshoff and H. F. Bettinger, Angew. Chem., Int. Ed., 2010, 49, 4125–4128 CrossRef PubMed; (c) B. Shen, J. Tatchen, E. Sanchez Garcia and H. F. Bettinger, Angew. Chem. DOI:10.1002/anie.201802197.
- (a) M. Bendikov, H. M. Duong, K. Starkey, K. N. Houk, E. A. Carter and F. Wudl, J. Am. Chem. Soc., 2004, 126, 7416–7417 CrossRef PubMed; (b) Y. Yang, E. R. Davidson and W. Yang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E5098–E5107 CrossRef PubMed.
- J. Hachmann, J. J. Dorando, M. Avilés and G. K.-L. Chan, J. Chem. Phys., 2007, 127, 134309 CrossRef PubMed.
- J. Krüger, N. Pavliček, J. M. Alonso, D. Pérez, E. Guitián, T. Lehmann, G. Cuniberti, A. Gourdon, G. Meyer, L. Gross, F. Moresco and D. Peña, ACS Nano, 2016, 10, 4538–4542 CrossRef PubMed.
- J. Krüger, F. Eisenhut, J. M. Alonso, T. Lehmann, E. Guitián, D. Pérez, D. Skidin, F. Gamaleja, D. A. Ryndyk, C. Joachim, D. Peña, F. Moresco and G. Cuniberti, Chem. Commun., 2017, 53, 1583–1586 RSC.
- J. I. Urgel, H. Hayashi, M. Di Giovannantonio, C. A. Pignedoli, S. Mishra, O. Deniz, M. Yamashita, T. Dienel, P. Ruffieux, H. Yamada and R. Fasel, J. Am. Chem. Soc., 2017, 139, 11658–11661 CrossRef PubMed.
- R. Zuzak, R. Dorel, M. Kolmer, M. Szymonski, S. Godlewski and A. M. Echavarren, Angew. Chem., Int. Ed. DOI:10.1002/anie.201802040.
- R. Zuzak, R. Dorel, M. Krawiec, B. Such, M. Kolmer, M. Szymonski, A. M. Echavarren and S. Godlewski, ACS Nano, 2017, 11, 9321–9329 CrossRef PubMed.
- J. Krüger, F. García, F. Eisenhut, D. Skidin, J. M. Alonso, E. Guitián, D. Pérez, G. Cuniberti, F. Moresco and D. Peña, Angew. Chem., Int. Ed., 2017, 56, 11945–11948 CrossRef PubMed.
- L. E. Dinca, C. Fu, J. M. MacLeod, J. Lipton-Duffin, J. L. Brusso, C. E. Szakacs, D. Ma, D. F. Perepichka and F. Rosei, ACS Nano, 2013, 7, 1652–1657 CrossRef PubMed.
- M. Zugermeier, M. Gruber, M. Schmid, B. P. Klein, L. Ruppenthal, P. Müller, R. Einholz, W. Hieringer, R. Berndt, H. F. Bettinger and J. M. Gottfried, Nanoscale, 2017, 9, 12461–12469 RSC.
- R. Dorel, P. R. McGonigal and A. M. Echavarren, Angew. Chem., Int. Ed., 2016, 55, 11120–11123 CrossRef PubMed.
- H. Walch, R. Gutzler, T. Sirtl, G. Eder and M. Lackinger, J. Phys. Chem. C, 2010, 114, 12604–12609 CrossRef.
- D. G. de Oteyza, A. García-Lekue, M. Vilas-Varela, N. Merino-Díez, E. Carbonell-Sanromà, M. Corso, G. Vasseur, C. Rogero, E. Guitián, J. I. Pascual, J. E. Ortega, Y. Wakayama and D. Peña, ACS Nano, 2016, 10, 9000–9008 CrossRef PubMed.
- (a) G. Galeotti, M. Di Giovannantonio, J. Lipton-Duffin, M. Ebrahimi, S. Tebi, A. Verdini, L. Floreano, Y. Fagot-Revurat, D. F. Perepichka, F. Rosei and G. Contini, Faraday Discuss., 2017, 204, 453–469 RSC; (b) N. Merino-Díez, J. Lobo-Checa, P. Nita, A. Garcia-Lekue, A. Basagni, G. Vasseur, F. Tiso, F. Sedona, P. K. Das, J. Fujii, I. Vobornik, M. Sambi, J. I. Pascual, J. E. Ortega and D. G. de Oteyza, J. Phys. Chem. Lett., 2018, 9, 2510–2517 CrossRef PubMed.
- S. W. Slayden and J. F. Liebman, Chem. Rev., 2001, 101, 1541–1566 CrossRef PubMed.
- A. Konishi, Y. Hirao, M. Nakano, A. Shimizu, E. Botek, B. Champagne, D. Shiomi, K. Sato, T. Takui, K. Matsumoto, H. Kurata and T. Kubo, J. Am. Chem. Soc., 2010, 132, 11021–11023 CrossRef PubMed.
- The exception is 6, whose additional Clar sextet with respect to 5 brings about an increased stability, a concomitant increase in its gap and thus a LUMO resonance at a higher energy.
- At this point, it is important to remark that the spectra of 2 showed a notable variability that remains out of the scope of this work and will be analyzed elsewhere.
- L. Gross, N. Moll, F. Mohn, A. Curioni, G. Meyer, F. Hanke and M. Persson, Phys. Rev. Lett., 2011, 107, 086101 CrossRef PubMed.
- G. Heimel, S. Duhm, I. Salzmann, A. Gerlach, A. Strozecka, J. Niederhausen, C. Bürker, T. Hosokai, I. Fernandez-Torrente, G. Schulze, S. Winkler, A. Wilke, R. Schlesinger, J. Frisch, B. Bröker, A. Vollmer, B. Detlefs, J. Pflaum, S. Kera, K. J. Franke, N. Ueno, J. I. Pascual, F. Schreiber and N. Koch, Nat. Chem., 2013, 5, 187–194 CrossRef PubMed.
- E. Goiri, P. Borghetti, A. El-Sayed, J. E. Ortega and D. G. de Oteyza, Adv. Mater., 2016, 28, 1340–1368 CrossRef PubMed.
- B. de la Torre, M. Švec, G. Foti, O. Krejčí, P. Hapala, A. Garcia-Lekue, T. Frederiksen, R. Zbořil, A. Arnau, H. Vázquez and P. Jelínek, Phys. Rev. Lett., 2017, 119, 166001 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc04402c |
| This journal is © The Royal Society of Chemistry 2018 |