
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReductive coupling of two aldehydes to unsymmetrical E-alkenes via phosphaalkene and phosphinate intermediates†
Juri
Mai
,
Anna I.
Arkhypchuk
,
Arvind Kumar
Gupta
 and
Sascha
Ott
*
and
Sascha
Ott
*
Department of Chemistry, Ångström Laboratory, Uppsala University, Box 523, 75120 Uppsala, Sweden. E-mail: sascha.ott@kemi.uu.se
First published on 11th June 2018
Abstract
Stilbenes with push–pull electronics are directly accessible from an electron-rich and an electron-deficient benzaldehyde in a novel reductive aldehyde cross-coupling reaction. The one-pot procedure is enabled by the oxidation of a transient phosphinite to the corresponding phosphinate which exhibits sufficient reactivity towards deactivated aldehydes.
Olefinations of carbonyl compounds such as aldehydes or ketones are one of the most fundamental conversions in organic synthesis.1 Many of the established procedures are classical name reactions such as the Wittig,2–6 Horner–Wadsworth–Emmons (HWE),7–9 Julia–Kocienski10,11 and Peterson olefinations.12–14 All these methodologies have in common that they make use of organo main group reagents to drive the olefination of one carbonyl compound. A direct conversion of two carbonyl compounds to alkenes is possible by the McMurry coupling where a low-valent titanium species acts as reducing agent and oxygen acceptor.15,16 This reaction is often described as “tricky”,17 requiring the in situ preparation of the reagent and extended heating in high-boiling solvents.18 Since the McMurry proceeds through a radical mechanism, functional groups that are prone to reductions are hardly compatible.19 Most importantly, however, the intermolecular coupling of two different carbonyl compounds yields a statistic mixture of the two symmetrical alkenes and the unsymmetrical product.20,21 Recently, we reported a new methodology for the selective reductive cross-coupling of two aldehydes to unsymmetrical E-alkenes.22 This new reaction makes use of an organophosphorus reagent, more specifically a phosphanylphosphonate 1-H,23 and achieves selectivity by the sequential addition of the two aldehyde substrates. The first aldehyde reacts with 1 to form a phosphaalkene intermediate 2 in a phosphorus version of the HWE reaction24,25 (Scheme 1, step 1). During this first step, the polarity of the carbon centre is inverted from δ+ to δ−, allowing the subsequent coupling chemistry to proceed through an ionic mechanism.‡
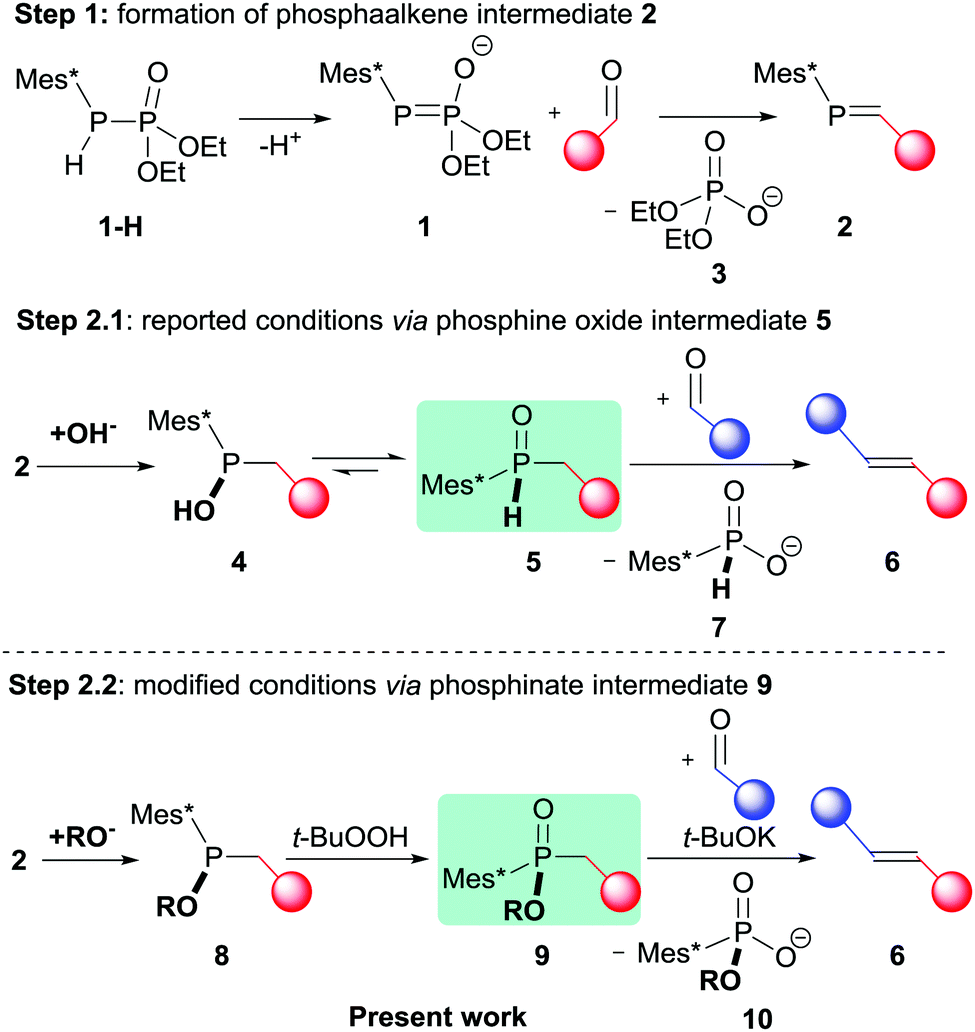 | ||
| Scheme 1 Reaction sequence for the reductive cross-coupling of two aldehydes to unsymmetrical E-alkenes via phosphine oxide intermediate 5 (step 2.1) and with modified reaction conditions via phosphinate intermediate 9 (step 2.2) R = Et or Me. | ||
Nucleophilic attack of added OH− on the P-centre of 2 affords a trivalent phosphinite species 4 that immediately tautomerizes to its pentavalent form 5. This phosphine oxide intermediate 5 is structurally analogous to Horner–Wittig reagents26 and reacts with a second aldehyde to form the olefinic product 6 under basic conditions.22 In comparison to the McMurry coupling, this new one-pot reaction works at room temperature within minutes. It has high selectivity for the formation of E-alkenes, and most importantly, it gives direct access to unsymmetrical alkenes from two non-identical aldehydes. While being an important proof of concept study, the substrate scope in the initial report was limited. For example, electron-rich stilbenes that would stem from benzaldehydes with electron-donating substituents remained elusive. Herein, we describe a significant improvement of the initial methodology that allows the use of such deactivated benzaldehydes in the second step of the reaction, thereby rendering push–pull trans-stilbenes directly accessible from two different aldehydes. Such types of stilbenes are important building blocks in organic electronics and photonic materials.27–29
In general, step 1 of the reductive coupling protocol depicted in Scheme 1 proceeds well for most aldehydes.23 The limitations in scope arise in step 2.1 of the one-pot procedure, where electron-donating C-substituents decrease the electrophilicity of the P-centre in the phosphaalkenes 2, rendering the latter unreactive to hydroxide attack. Even if phosphine oxide 5 is formed, the electron-rich substituents lower the acidity of the α-proton, making electron-rich aldehydes generally unsuitable to be used as the first substrate of the sequence. In contrast, phosphine oxides 5 with electron-withdrawing C-substituents are readily deprotonated and react with a larger variety of aldehydes. The scope for the second aldehyde is however also limited and electron-rich substituents in the second aldehyde render them too unreactive for the nucleophilic attack of deprotonated phosphine oxide 5, and decomposition pathways become dominating.
With this background knowledge, we decided to modify the sequence with the goal to increase the reactivity of phosphine oxide 5. We hypothesized that the presence of more oxygen substituents at the P-centre should have exactly this effect, as they will increase the acidity of the α-protons while increasing the nucleophilicity of the deprotonated form. In fact, the same rational has led to the development of the classical HWE reagents about 60 years ago.7,30 We therefore targeted a sequence in which phosphaalkene 2 is first converted to a phosphinite 8, followed by oxidation to phosphinate 9 (Scheme 1, step 2.2.). According to the rational outlined above, the deprotonated form of 9 is envisaged to be more reactive than deprotonated phosphine oxide 5. Initial support for this hypothesis was obtained from DFT calculations at the B3LYP level of theory with a 6-311G(d,p) basis set (see ESI,† for details), which showed a higher Mulliken charge at the P-centre in 9 (+1.246) compared to that in 5 (+0.986).
Addition of methanol across the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in 2 can be accomplished by the addition of methanolic solution of tetrabutylammonium methoxide (TBAOMe/MeOH), and can conveniently be monitored by 31P-NMR spectroscopy. In a comparative study, it can be seen that the rate for this addition strongly depends on the electronic nature of the substituent R1. As shown in Fig. 1, the consumption of the phosphaalkenes with electron-withdrawing substituents (R1 = CN or Br) and the formation of phosphinites 8 proceeds smoothly within minutes after TBAOMe addition. Neutral phosphaalkens (R1 = H) are converted within one hour, while electron-rich phosphaalkenes react more sluggishly. This reactivity trend is thus similar to that observed for hydroxide attack at phosphaalkenes in step 2.1, and the reason for using electron-deficient aldehydes in the first step of the synthetic sequence. Conversion of phosphinite 8 to phosphinate 9 can be achieved by a variety of oxidants,31 with t-BuOOH giving the most satisfying results. An aqueous hydrogen peroxide solution can also be used, however, has been found to hydrolyze substituents such as cyanides that may be present in the compound.32 Under these conditions, a crystal structure of phosphinate 9 could be obtained (Fig. 2). In comparison to the crystal structure of 5 (R1 = CN, see ESI†), a shortening of the bond by 0.025 Å between phosphorus and the α-carbon atoms is observed (P–Cα bond length in 9
C bond in 2 can be accomplished by the addition of methanolic solution of tetrabutylammonium methoxide (TBAOMe/MeOH), and can conveniently be monitored by 31P-NMR spectroscopy. In a comparative study, it can be seen that the rate for this addition strongly depends on the electronic nature of the substituent R1. As shown in Fig. 1, the consumption of the phosphaalkenes with electron-withdrawing substituents (R1 = CN or Br) and the formation of phosphinites 8 proceeds smoothly within minutes after TBAOMe addition. Neutral phosphaalkens (R1 = H) are converted within one hour, while electron-rich phosphaalkenes react more sluggishly. This reactivity trend is thus similar to that observed for hydroxide attack at phosphaalkenes in step 2.1, and the reason for using electron-deficient aldehydes in the first step of the synthetic sequence. Conversion of phosphinite 8 to phosphinate 9 can be achieved by a variety of oxidants,31 with t-BuOOH giving the most satisfying results. An aqueous hydrogen peroxide solution can also be used, however, has been found to hydrolyze substituents such as cyanides that may be present in the compound.32 Under these conditions, a crystal structure of phosphinate 9 could be obtained (Fig. 2). In comparison to the crystal structure of 5 (R1 = CN, see ESI†), a shortening of the bond by 0.025 Å between phosphorus and the α-carbon atoms is observed (P–Cα bond length in 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.810(5) Å; in 5:1.835(2)). This observation is in agreement with a more polar character of the P–C bond that results from the alkoxy substituent at the phosphorus centre in compound 9.
:1.810(5) Å; in 5:1.835(2)). This observation is in agreement with a more polar character of the P–C bond that results from the alkoxy substituent at the phosphorus centre in compound 9.
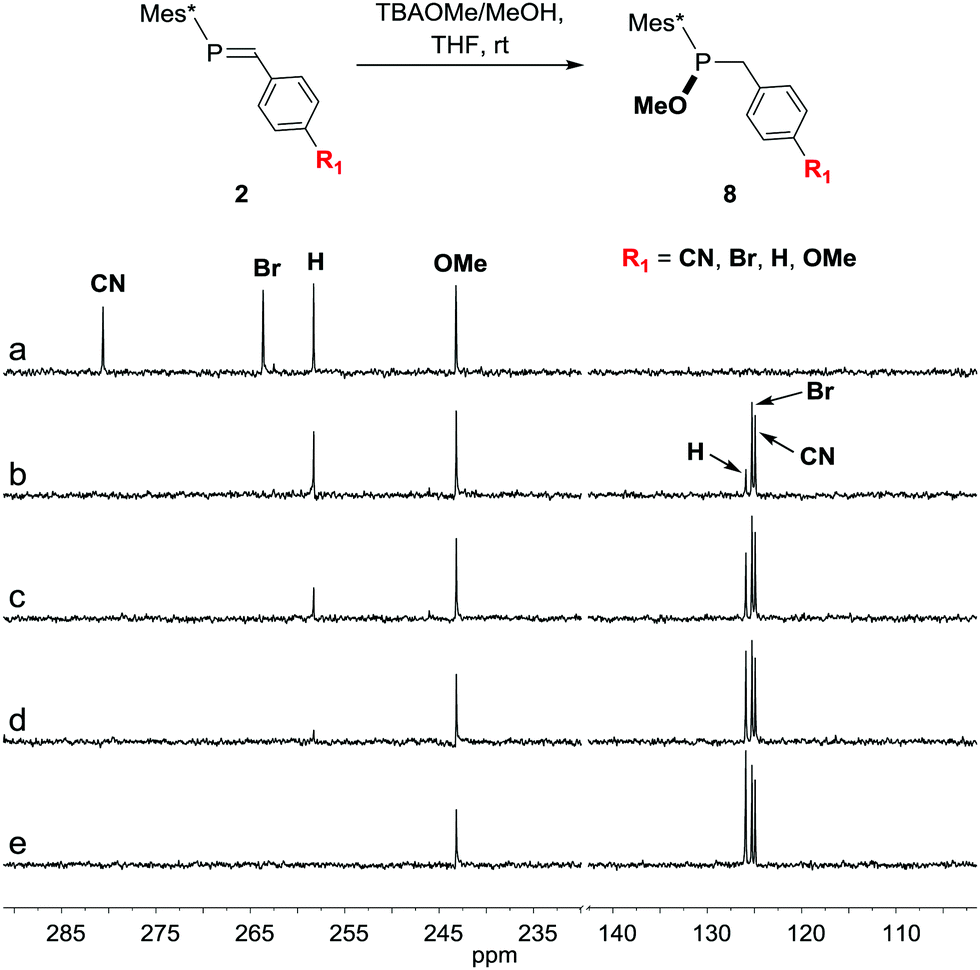 | ||
| Fig. 1 31P NMR spectroscopic monitoring of the addition of a methanolic tetrabutylammonium methoxide (TBAOMe/MeOH) solution to a mixture of phosphaalkenes with electron-withdrawing (R1 = CN, Br), neutral (H) and electron-donating (OMe) substituents. (a) Reaction mixture of isolated phosphaalkene starting materials. (b) 7 minutes, (c) 22 minutes, (d) 53 minutes and (e) 2.5 hours after TBAOMe/MeOH addition. | ||
 | ||
| Fig. 2 ORTEP plot of phosphinate 9 (with R = Et and R1 = C(O)NH2) at 50% probability ellipsoids. Hydrogen atoms are omitted for clarity. The crystal was isolated from oxidation of 8 (R = Et and R1 = CN) with aqueous H2O2 solution. Selected bond lengths [Å] and angles [°]: P1–C2 1.810(5), P1–C10 1.826(4), P1–O1 1.478(3), P1–O2 1.579(4), C2–P1–C10 103.5(2), O1–P1–O2 112.2(2). | ||
With a reliable procedure to phosphinates 9 (R1 = CN or Br) in hand, their reactivity towards deactivated, electron-rich aldehydes was explored. This step of the sequence has precedence in the literature, and was reported by Horner in 1961.33 Much to our satisfaction, the reaction of 9 with electron-rich benzaldehydes such as those with 4-methoxy- or 4-morpholino-substituents in the presence of KOtBu proceeds well, and the unsymmetrical push–pull stilbenes are formed without observation of any symmetric stilbenes (Table 1). The reaction is highly selective for the formation of the E-isomer, with only trace amounts (1–5%) of Z-isomer being observed in some instances (see Table S1 in ESI†). This reactivity is vastly different to that of 5 which did not afford any alkenes with deactivated benzaldehydes.
| Entry | 1st aldehyde | 2nd aldehyde | Product isolated yield (crude yield) |
|---|---|---|---|
| a Coupling was performed with 0.5 equivalent of the 2nd aldehyde. | |||
| 1 |

|

|
|
| 2 |
|

|

|
| 2′ |

|
|
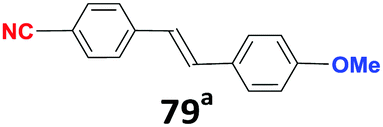
|
| 3 |

|

|

|
| 3′ |
|

|

|
| 4 |
|

|

|
| 5 |

|

|

|
| 6 |

|

|

|
| 7 |

|

|

|
The yields for the overall reductive coupling of the two aldehydes to the unsymmetric stilbenes are decent to good, and similar to those often reported for the olefination of unreactive aldehydes in classical HWE reactions.9 The advantage of our protocol is that the preparation of phosphonate reagents that are needed for HWE chemistry and usually formed in 2–3 steps from commercial starting materials is avoided in our direct coupling methodology. In case the second aldehyde is economically valuable, it can be used as the limiting reagent in the one-pot sequence. Thus, addition of 0.5 equivalents (relative to 1) of second aldehyde increases the overall yield of the coupling to 70–80% isolated yield (entries 2′ and 3′ in Table 1). Noteworthy is also the coupling of 4-cyano- with 4-nitrobenzaldehyde to the unsymmetric stilbene in 67% yield (entry 7), as neither of the substrates are compatible with the radical mechanism of the McMurry coupling.19,34
As mentioned above, 31P NMR spectroscopy is a convenient tool to follow all steps of the sequence. As shown in Fig. 3, phosphaalkene 2 formation from the first aldehyde and phosphanylphosphonate 1-H under basic conditions (via deprotonated 1) proceeds smoothly. Addition of TBAOMe/MeOH converts 2 to phosphinite 8 which features at a 31P NMR resonance of about 125 ppm. As expected, oxidation leads to an additional upfield shift and phosphinate 9 can be observed at ∼45 ppm. The acid/base equilibrium is lying rather on the side of the protonated form for 9, as the proton-coupled 31P NMR spectra (see ESI†) indicate couplings to two methylene protons in α-position to the respective P-centres. This finding is consistent with the experimental observation that additional KtOBu needs to be added to drive the reaction of 9 with a second aldehyde to the desired alkene and the phosphonate by-product 10. Compound 10, being a valuable organophosphorus compound, may be recovered in procedures similar to those developed for the reductive recycling of phosphine oxides, phosphinates and phosphonates to phosphines.35,36 Thus, 10 can in principle be reduced to the corresponding Mes*PH2 and reused for the preparation of 1-H.
 | ||
| Fig. 3 31P NMR spectroscopic investigations of individual reaction steps. Deprotonation of phosphanylphosphonate 1-H to 1 (a → b), followed by the phospha-HWE reaction (b → c) to form phosphaalkene 2 and the diethyl phosphate by-product 3. Further conversion to the phosphinite intermediate 8 (c → d) which is oxidized to phosphinate 9 (d → e). In the final step (e → f), 9 is reacted with a second aldehyde to give the alkene product and the Mes*-phosphonate by-product 10 (R1 = 4-PhCN). | ||
In summary, we could show that the one-pot reductive cross-coupling of aldehydes to alkenes can be extended to allow the use of deactivated aldehydes in the second step of the protocol. This reactivity is achieved by a modification of the original protocol that increases the amount of oxygen substituents at the P-centre. The thereby obtained phosphinate 9 is more reactive than the previously reported phosphine oxide 5, and allows the formation of trans-stilbenes with push–pull electronic properties directly from two aldehydes. The developed methodology is advantageous over the McMurry reaction from a selectivity viewpoint, but also compared to Wittig or HWE-type chemistry that is usually used for the preparation of stilbenes. The advantage comes from the fact that our procedure is a one-pot reaction, and thus omits the necessity to synthesize bromide and ylid/phosphonate precursors that are typically needed in the three steps of a typical Wittig sequence.
The authors wish to acknowledge Dr Andreas Orthaber for valuable discussions, and the Swedish Research Council for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Modern Carbonyl Olefination, ed. T. Takeda, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2004 Search PubMed.
- G. Wittig and G. Geissler, Liebigs Ann. Chem., 1953, 580, 44 CrossRef.
- G. Wittig and U. Schöllkopf, Chem. Ber., 1954, 87, 1318 CrossRef.
- B. E. Maryanoff and A. B. Reitz, Chem. Rev., 1989, 89, 863 CrossRef.
- O. I. Kolodiazhnyi, Phosphorus Ylides, Wiley-VCH Verlag GmbH, Weinheim, 2007, p. 359 Search PubMed.
- G. Wittig, Science, 1980, 210, 600 Search PubMed.
- W. S. Wadsworth and W. D. Emmons, J. Am. Chem. Soc., 1961, 83, 1733 CrossRef.
- J. Boutagy and R. Thomas, Chem. Rev., 1974, 74, 87 CrossRef.
- W. S. Wadsworth, Organic Reactions, John Wiley & Sons, Inc., 2005, p. 73 Search PubMed.
- M. Julia and J.-M. Paris, Tetrahedron Lett., 1973, 14, 4833 CrossRef.
- P. J. Kocienski, B. Lythgoe and S. Ruston, J. Chem. Soc., Perkin Trans., 1978, 1, 829 RSC.
- D. J. Peterson, J. Org. Chem., 1968, 33, 780 CrossRef.
- L. F. v. Staden, D. Gravestock and D. J. Ager, Chem. Soc. Rev., 2002, 31, 195 RSC.
- Z. Wang, Comprehensive Organic Name Reactions and Reagents, John Wiley & Sons, Inc., 2010 Search PubMed.
- J. E. McMurry and M. P. Fleming, J. Am. Chem. Soc., 1974, 96, 4708 CrossRef.
- J. E. McMurry, Chem. Rev., 1989, 89, 1513 CrossRef.
- L. Kürti and B. Czakó, Strategic Applications of Named Reactions in Organic Synthesis, Elsevier Academic Press, San Diego, 2005, pp. 276–277 Search PubMed.
- M. Ephritikhine, Chem. Commun., 1998, 2549 RSC.
- J. E. McMurry, Acc. Chem. Res., 1974, 7, 281 CrossRef.
- J. E. McMurry, M. P. Fleming, K. L. Kees and L. R. Krepski, J. Org. Chem., 1978, 43, 3255 CrossRef.
- J. E. McMurry and L. R. Krepski, J. Org. Chem., 1976, 41, 3929 CrossRef.
- K. Esfandiarfard, J. Mai and S. Ott, J. Am. Chem. Soc., 2017, 139, 2940 CrossRef PubMed.
- K. Esfandiarfard, A. I. Arkhypchuk, A. Orthaber and S. Ott, Dalton Trans., 2016, 45, 2201 RSC.
- A. Marinetti and F. Mathey, Angew. Chem., Int. Ed., 1988, 27, 1382 CrossRef.
- A. I. Arkhypchuk, Y. V. Svyaschenko, A. Orthaber and S. Ott, Angew. Chem., Int. Ed., 2013, 52, 6484 CrossRef PubMed.
- J. Clayden and S. Warren, Angew. Chem., Int. Ed., 1996, 35, 241 CrossRef.
- S. R. Marder, D. N. Beraten and L.-T. Cheng, Science, 1991, 252, 103 Search PubMed.
- F. Bureš, RSC Adv., 2014, 4, 58826 RSC.
- M. Klikar, P. Solanke, J. Tydlitát and F. Bureš, Chem. Rec., 2016, 16, 1886 CrossRef PubMed.
- L. Horner, H. Hoffmann, H. G. Wippel and G. Klahre, Chem. Ber., 1959, 92, 2499 CrossRef.
- T. A. Van der Knaap, T. C. Klebach, R. Lourens, M. Vos and F. Bickelhaupt, J. Am. Chem. Soc., 1983, 105, 4026 CrossRef.
- J. E. McIsaac, R. E. Ball and E. J. Behrman, J. Org. Chem., 1971, 36, 3048 CrossRef.
- L. Horner, H. Hoffmann, W. Klink, H. Ertel and V. G. Toscano, Chem. Ber., 1961, 95, 581 CrossRef.
- R. Dams, M. Malinowski and H. J. Geise, Recl. Trav. Chim. Pays-Bas, 1982, 101, 112 CrossRef.
- D. Herault, D. H. Nguyen, D. Nuel and G. Buono, Chem. Soc. Rev., 2015, 44, 2508 RSC.
- M. L. Clarke and J. M. J. Williams, in Organophosphorus Reagents. A Practical Approach in Chemistry, ed. P. J. Murphy, Oxford University Press, 2004, p. 27 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and characterization of all compounds. CCDC 1832099 and 1832100. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc04218g |
| ‡ Phosphanylphosphonate 1-H was dissolved in dry THF and LDA (1.15 eq. solution in THF) was added at room temperature. The first aldehyde (1.05 eq.) was added, and the formation of the phosphaalkene was monitored by 31P NMR. After complete transformation, typically within a few minutes, tetrabutylammonium methoxide solution (1 eq., 20% TBAOMe in MeOH) was added, and the mixture was stirred at room temperature for 30 minutes. After full conversion (followed by 31P NMR) t-BuOOH (1 eq., 14% weight solution in benzene) was added at room temperature. The oxidation was performed open to air and was complete in 5–6 hours. KOtBu (3 eq.) was added simultaneously with the second aldehyde, and the mixture was stirred at room temperature. Conversion of the phosphinate intermediate 9 to the final phosphonate by-product 10 was monitored by 31P NMR. After completion of the coupling step, typically within 15–90 minutes, the reaction was quenched with a saturated aqueous solution of NH4Cl. The aqueous phase was extracted with ethyl acetate, and the combined organic phases were dried over MgSO4. Evaporation of the solvents under reduced pressure afforded the crude products, which were purified by silica gel column chromatography. Crystallographic data of a phosphine oxide 5 (R1 = CN, see ESI†) and a phosphinate 9 (see Fig. 2) has been deposited at the CCDC under 1832099 and 1832100, respectively.† |
| This journal is © The Royal Society of Chemistry 2018 |