
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlpha-helical folding of SilE models upon Ag(His)(Met) motif formation†
Valentin
Chabert
 a,
Maggy
Hologne
b,
Olivier
Sénèque
c,
Olivier
Walker
b and
Katharina M.
Fromm
*a
a,
Maggy
Hologne
b,
Olivier
Sénèque
c,
Olivier
Walker
b and
Katharina M.
Fromm
*a
aUniversity of Fribourg, Department of Chemistry, Chemin du Musée 9, 1700 Fribourg, Switzerland. E-mail: katharina.fromm@unifr.ch
bUniversité de Lyon, CNRS, UCB Lyon 1, ENS-Lyon, Institut des Sciences Analytiques, UMR 5280, 5 rue de la Doua, 69100 Villeurbanne, France
cUniversité Grenoble Alpes, CNRS, CEA, BIG/LCBM (UMR 5249), 38000 Grenoble, France
First published on 22nd August 2018
Abstract
The SilE protein is suspected to have a prominent role in Ag+ detoxification of silver resistant bacteria. Using model peptides, we elucidated both qualitative and quantitative aspects of the Ag+-induced α-helical structuring role of His- and Met-rich sequences of SilE, improving our understanding of its function within the Sil system.
The bactericidal power of Ag+ has been exploited for hundreds of years, and recent efforts have been made to develop new silver-based compounds able to tackle multidrug-resistant bacteria.1,2 However, several Gram-negative bacteria are able to survive in silver contaminated media, thanks to intrinsic and/or acquired HME-RND (Heavy-Metal-Efflux Resistance-Nodulation-Division) efflux pumps.3,4 The latter are the most common bacterial defence against toxic metal ions.5 The Cus and Sil systems are part of the HME-RND family and have shown the capacity to make bacteria silver resistant.6–9 While the Cus system is expressed in the context of chromosomal mutations, the Sil system is plasmid encoded. Besides the fact that they can be horizontally transferred, resistance plasmids have relatively low fitness costs in comparison with chromosomal resistance mutations.10 As a consequence, plasmid-encoded resistances are deemed to be more widespread than mutational resistances.11 Hence, the full understanding of the Sil system is of prime importance in the context of fighting silver resistant bacteria. Both systems function along similar lines and possess a common basis (CusCFBA and SilCFBA). However, this basis is not efficient enough to provide a significant silver tolerance, and needs to be complemented by a control of the cellular silver concentration. Therefore, the CusCFBA transporter is associated to a deficiency in OmpC and OmpF porins, and the SilCFBA transporter is associated to three other proteins, SilP, SilE and SilG, of which only SilE is mandatory to confer the resistance.7 Moreover, Randall et al. have shown that SilE could substitute the need for porin loss associated with the Cus system.7 All this points out the prominent role of SilE in the bacterial resistance to Ag+. Nevertheless, the structure of the latter has never been solved and its mode of action is still debated. Previous studies have proposed histidine and methionine residues to be engaged in the coordination of Ag+ ions by SilE via eight MX2H and HX2M motifs and one HXM motif, all individually able to bind a single Ag+ ion with Kd in the μM range.12–14 However, the involvement of these motifs in Ag+ coordination in the full-length SilE protein has never been proved, and Ag+/SilE binding affinities have not been reported to date. The mechanistic aspects of the Ag+-induced folding of the protein and its structural organization remained also unclear, making the understanding of its structure/function relationship difficult. Here we report qualitative and quantitative aspects of the Ag+ coordination by structural motifs of SilE, through the characterization of the silver binding sites in terms of coordination numbers and geometries, binding affinities, and Ag+-induced structural folding. Based on our studies, the first structures of SilE key sequences have been solved and a non-cooperative Ag+ binding is proposed for SilE.
The eight MX2H and HX2M motifs of SilE form two types of “twin” sequences, namely MX2HX6HX2M (type A) and HX2MX3HX2M (type B) (Fig. 1). Model peptides of these four sequences have been synthesized as N-terminal acetylated and C-terminal amidated peptides. The B1 motif has been additionally studied as 14-amino acid peptide (B1b), to investigate the potential involvement of Met91 in Ag+ coordination, and a sixth model peptide corresponding to the 28 C-terminal amino acids of SilE (B2b) has also been studied to examine the Ag+ coordination of a model peptide containing three HX2M motifs. The different peptides were investigated by 1H NMR titrations to determine their Ag+-binding capacity. The 1H chemical shifts of both His and both Met residues (His-Hδ2, His-Hε1 and Met-Hε) are significantly affected by the addition of Ag+, indicating their involvement in Ag+ coordination. The evolution of the chemical shift is clearly in agreement with the binding of two Ag+ ions per peptide with a plateau after 2 equiv. of Ag+ added (Fig. 2a and Fig. S1, ESI†). Similarly, the B2b model that harbors three HX2M motifs can bind up to three silver ions (Fig. S1, ESI†). The behavior of B1b, which displays two His and three Met, is more complicated. It is described in detail in the ESI† (Fig. S1 and S2). This peptide can bind three Ag+ ions, the third of which has a lower affinity (mM range vs. μM, see below and ESI†). The monitoring of the chemical shifts reveals that the HX2M motifs are the two primary binding sites. The third Ag+ ion may bind to Met90 and Met91. For all peptides, DOSY experiments were recorded in the presence or absence of Ag+ ions (Fig. S3–S8, ESI†) in order to compare the diffusion coefficients of the apo-peptides and their silver complexes. They indicate species of similar size suggesting the formation of silver complex species involving a single peptide, i.e. Ag2P or Ag3P. Thus, we can conclude that all these peptides bind one Ag+ ion per MX2H or HX2M motif. From previous studies on HX2M and MX2H tetrapeptides, we can infer a His–Ag–Met coordination mode, but at this stage, His–Ag–His and Met–Ag–Met coordination modes cannot be ruled out. Further structural details about the Ag+/SilE interaction arise from circular dichroism (CD) and NMR. The two types of sequences (MX2HX6HX2M, A and HX2MX3HX2M, B), which exhibit random-coil conformations in their apo-form, adopt different structures in the presence of silver ions. CD experiments reveal the appearance of two minima at 207 nm and 223 nm for type B peptides, clearly indicating that they are folding into α-helices upon Ag+ addition (Fig. 2b), in contrast to type A peptides, which exhibit a poor α-helix signature evolution during Ag+ complexation and appear thus less structured in their holo-form (Fig. S9, ESI†).15
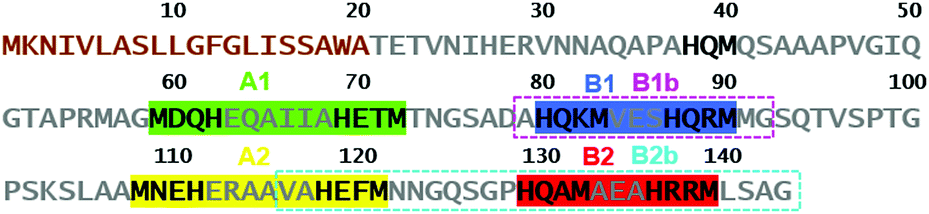 | ||
| Fig. 1 Amino acid sequence of SilE. The first twenty amino acids (brown) are cleaved after the periplasm targeting. The His- and Met- containing regions hold two MX2HX6HX2M motifs (A1 and A2) and two HX2MX3HX2M motifs (B1 and B2) that are studied as structural model peptides of SilE. B1b and B2b have been additionally studied as longer model peptides of B1 and B2 sequences, respectively. | ||
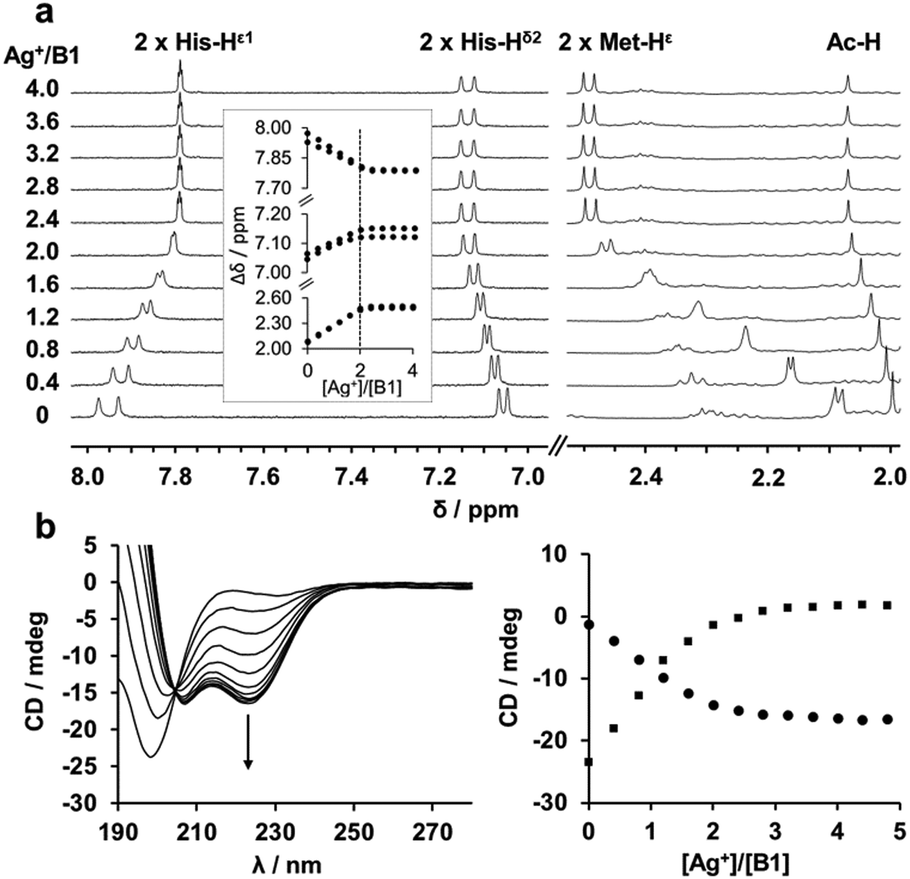 | ||
Fig. 2
B1/Ag+ NMR and CD titrations. (a) Histidine (His-Hε1 and His-Hδ2) and methionine (Met-Hε) 1H resonances shift by addition of AgClO4 (0 to 4 mM) to a solution of B1 (1 mM) in HEPES buffer (20 mM, pD 7.8). The insert depicts the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry of the Ag2B1 complex. (b) α-Helix folding of B1 (10 μM, in NH4Ac 1 mM, pH 7.4) by addition of AgClO4 (0 to 48 μM) evidenced by the increase of the signature at 223 nm (left). Plot of the CD signal at 199 nm (square) and 223 nm (circle). :1 stoichiometry of the Ag2B1 complex. (b) α-Helix folding of B1 (10 μM, in NH4Ac 1 mM, pH 7.4) by addition of AgClO4 (0 to 48 μM) evidenced by the increase of the signature at 223 nm (left). Plot of the CD signal at 199 nm (square) and 223 nm (circle). | ||
To gain deeper understanding of the coordination geometries and the structural features of the silver binding sequences of SilE, the NMR solution structures of the different Ag+/SilE-derived peptide complexes have been solved (Fig. 3). As expected, the Ag+ binding to B1 and B2 induces a well-defined helical structure of the respective model peptides. On the other hand, A1 and A2 remain partly unstructured. In both cases, however, the orientation of His and Met side chains, which are alternating on the same side of the α-helix in type B peptides, clearly indicates that each Ag+ ion is bound to one HX2M or MX2H motif, excluding His–Ag–His or Met–Ag–Met coordination geometries. Therefore, the 1:1 Ag+/HX2M or MX2H stoichiometry pointed out by both tetra- and poly-peptide model studies suggests a linear His–Ag–Met coordination mode, which is common for Ag+ ions. Moreover, B1b and B2b peptides adopt an α-helical fold upon Ag+ binding similar to those of B1 and B2, respectively, and their structures overlap to a large extent (Fig. S12, ESI†). In the case of B1b, the potential coordination of a third Ag+ ion by Met90 and Met91 (see above and ESI†) does not influence the folding of B1b. In the case of B2b, no NOE between the N-terminal HEFM motif and the two C-terminal motifs composing the B2 sequence are observed, indicating that they bind different Ag+ ions.
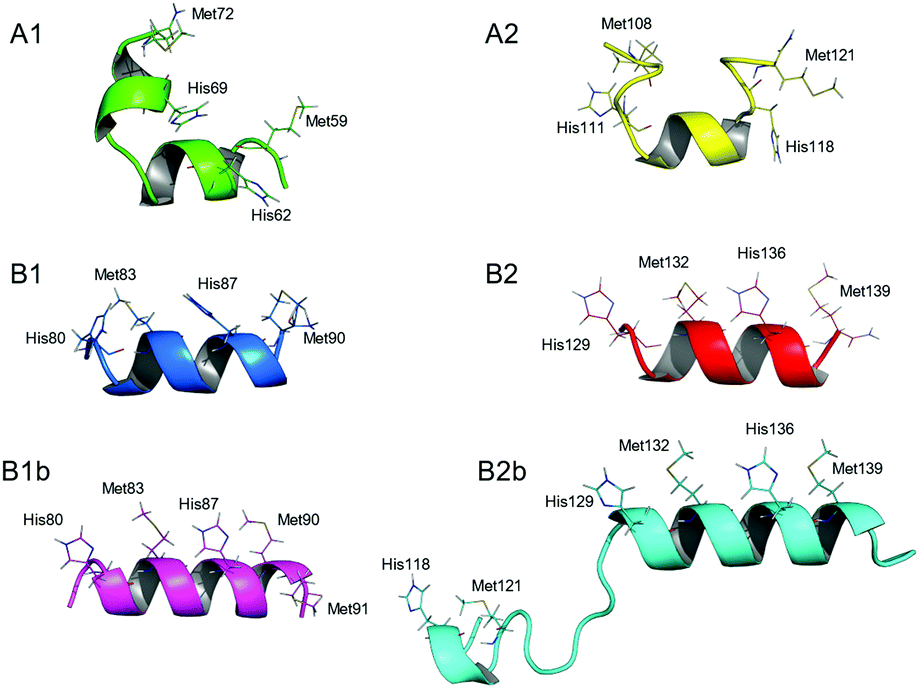 | ||
| Fig. 3 Lowest energy NMR solution structure of the six SilE-derived peptides in presence of Ag+. His and Met residues are represented by lines. Type B peptides fold into two α-helices stretching from residues Gln81 to Arg89 and Gln130 to Arg138 whereas type A peptides own less defined secondary structure. | ||
As previously shown, isolated MX2H and HX2M motifs intrinsically bind silver ions with moderately strong affinities (logKass = 5.3–6.6).13 However, it has to be determined whether the proximity of the binding sites and the Ag+-induced α-helix folding of the backbone affect the silver binding affinities of the various MX2H or HX2M motifs. Fluorescence competition titrations were chosen to determine the binding constants of the model peptides, requiring the design of a fluorescent probe. Inspired by the MX2H and HX2M motifs of SilE, a tryptophan containing tetrapeptide (HEWM) has been synthesized for this purpose, and its ability to bind Ag+ has been investigated (Fig. 4). A fluorescence titration of HEWM by Ag+ in HEPES buffer shows the formation of a 1:1 complex with a 50% quenching of the tryptophan emission upon Ag+ binding. 1H NMR confirms the stoichiometry of the complex and the binding of both histidine and methionine to Ag+ (Fig. S13, ESI†).13 Based on the difference of fluorescence intensity between the apo- and holo-forms of the probe, competition experiments were performed with four different tetrapeptide competitors of known Ag+ affinity (MDQH, MNEH, HEFM and HQAM)13 in order to determine the association constant of the probe (Kass = [AgP]/[Ag][P]), yielding logKass = 6.4 ± 0.2 (Fig. 4, Fig. S14–S16 and Table S3, ESI†).16 Then, in order to determine the binding affinities of A and B peptides, similar experiments were performed with our six SilE-derived peptides using HEWM as competitive fluorescent probe (Fig. S17–S20 and Table S4, ESI†). The resulting binding constants are in the same order of magnitude as those obtained for the corresponding tetrapeptide complexes. For instance, the peptide A1 binds two Ag+ (K1 = [AgP]/[Ag][P] and K2 = [Ag2P]/[Ag][AgP]) with logK1 = 6.6 ± 0.3 and logK2 = 5.6 ± 0.4, while the two individual motifs composing the sequence, HETM and MDQH, bind one Ag+ with logK = 6.4 ± 0.1 and 5.8 ± 0.1, respectively, when studied as tetrapeptides (Table 1). However, affinity constants of the trimetallic Ag3B1b and Ag3B2b complexes could not be extracted with this method, since it ends up with a too large standard deviation on the K3 value. Overall, with stepwise association constants (logK1 and logK2) between 5.1 and 6.7 (±0.5), the affinities of the herein described models for the two silver ions are in the same range as the intrinsic affinities of the MX2H and HX2M sites (logKass = 5.3–6.6).13 Therefore, these similarities confirm the His–Ag–Met coordination mode in type A and B peptides. Moreover, the binding affinities of B1 and B2 being similar to those of A1 and A2, no significant effect of the peptide structuration has been observed. Obviously, the presence of two silver binding sites in the model and the Ag+-induced α-helix folding of the peptide do not have a significant effect on the silver binding constants. Therefore, the four MX2HX6HX2M and HX2MX3HX2M sequences found in SilE are proposed to bind Ag+ in a non-cooperative binding mode.
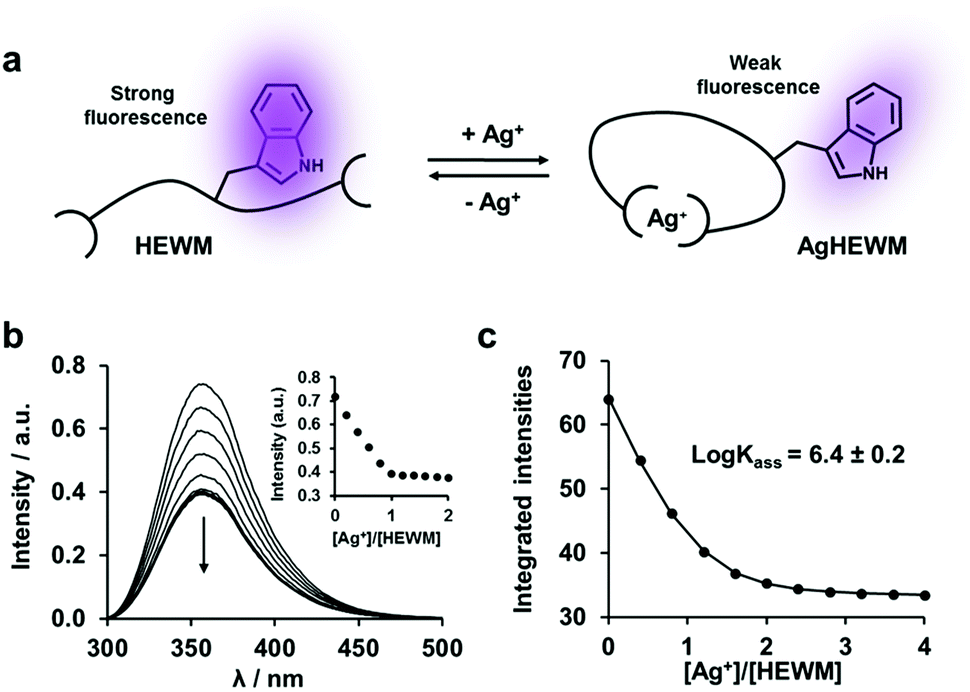 | ||
| Fig. 4 Characterization of the fluorescent probe. (a) Principle of the probe. (b) Spectrofluorimetric titration of HEWM (200 μM) by AgClO4 (0 to 400 μM) in HEPES buffer (20 mM, pH 7.4), based on the tryptophan fluorescence (λex: 280 nm) quench induced by Ag+ complexation. Insert depicts the 1:1 stoichiometry of the complex. (c) Plot of the variation of fluorescence integrated intensities by addition of AgClO4 (0 to 40 μM) to a solution of HEWM (10 μM) in competition with MDQH (10 μM), in a HEPES buffer (4 mM). The solid line corresponds to the fit obtained with Dynafit,16 which yielded logKass = 6.4 ± 0.2. | ||
Kass) of 1:1 and 2:1 Ag+/peptide complexes
| Model | logKassa |
Model | logKassb |
|---|---|---|---|
|
a logKass of HX2M and MX2H motifs studied as tetrapeptides from ref. 13.
b logKass of the model peptides determined in the herein described study.
|
|||
| HQKM | 5.7 ± 0.1 | B1 | logK1 = 6.2 ± 0.3 |
| HQRM | 5.5 ± 0.1 | B1 | logK2 = 5.1 ± 0.5 |
| HQAM | 5.9 ± 0.1 | B2 | logK1 = 6.5 ± 0.3 |
| HRRM | 5.3 ± 0.1 | B2 | logK2 = 5.3 ± 0.4 |
| HETM | 6.4 ± 0.1 | A1 | logK1 = 6.6 ± 0.3 |
| MDQH | 5.8 ± 0.1 | A1 | logK2 = 5.6 ± 0.4 |
| HEFM | 6.6 ± 0.1 | A2 | logK1 = 6.7 ± 0.4 |
| MNEH | 5.4 ± 0.1 | A2 | logK2 = 5.5 ± 0.3 |
In the context of accurate characterization of protein–metal interactions, the use of model peptides can help to reach qualitative and quantitative insights, which are not necessarily attainable by working with an entire protein. In this instance, while several research groups have investigated the role of SilE for two decades, no structural or numerical characterization of the metal centers has arisen. In contrast, the herein described work not only provides the structure of the four silver binding sequences of SilE, but also quantises the silver docking. As suggested by previous studies on MX2H and HX2M ultrashort model peptides of SilE, the latter seems to bind up to 8 Ag+ ions via its MX2HX6HX2M and HX2MX3HX2M sequences, each of them binding two Ag+ ions. A ninth Ag+ ion could nevertheless be bound to the isolated HXM motif (H38–M40) with a similar affinity.13 Asiani et al. suggested a complete folding of SilE after the binding of 6 Ag+ ions (out of a total of 8 Ag+ ions bound), and the presence of two core motifs (A77–M91 and E110–F120) which, when folded into α-helices, should facilitate the folding of the rest of the protein.17 The different backbone foldings between type A and type B sequences upon Ag+ complexation support this hypothesis. However, while the first core motif corresponds to the herein described B1 peptide, which folds into a stable α-helical structure upon Ag+ binding, the second core motif is a truncated version of the A2 peptide, which does not adopt a well folded secondary structure in the presence of Ag+ (Fig. 3 and Fig. S9, ESI†). Furthermore, contrary to previous assumptions from Asiani et al. who assumed that each single α-helix is unable to bind Ag+ by itself, each type A and type B sequence individually binds two Ag+ ions, even when histidine and methionine side chains are on the same side of an α-helix (type B), as determined by 1H NMR.17 Of particular interest is this alternation of histidine and methionine residues on the same face of the α-helix in B1 and B2 peptides that clearly suggests the formation of His–Ag–Met motifs with linear geometry, which is common in Ag+ coordination chemistry. In order to assess if these peptides can accommodate linear His–Ag–Met coordination, the structure of B2 was calculated once again using NMR-derived distance and dihedral angle restraints but forcing a linear His·N–Ag–S·Met geometry.18 The obtained structure retains the helical fold of B2 and displays no violation of NOE and dihedral constraints. It clearly establishes that this linear His·N–Ag–S·Met geometry is indeed possible within a HX2M motif as part of an α-helix (Fig. 5).
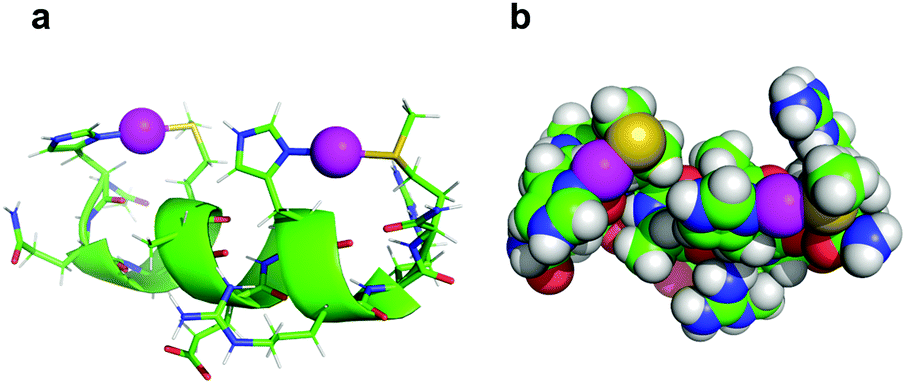 | ||
| Fig. 5 Model of the coordination mode of two Ag+ by B2. The spacing of two amino acids between the histidine and methionine residues of the two HX2M binding sites brings the two ligands onto the same side of the α-helix, allowing a linear coordination of each Ag+ ion (purple spheres). The His·Nδ1–Ag+ and MetSδ–Ag+ distances used in the calculation were 2.1 and 2.5 Å respectively.18 | ||
The plots of NMR and CD titrations of the herein described model peptides by Ag+ always adopt hyperbolic binding curves, suggesting a non-cooperative binding of Ag+ ions by the different binding sequences of SilE. Numerical values of stepwise affinity constants of the Ag+/SilE-derived peptide complexes (6.2 ± 0.3 < logK1 < 6.7 ± 0.4 and 5.1 ± 0.5 < logK2 < 5.6 ± 0.4) support this hypothesis. Most surprising, however, is that type A and type B peptides bind Ag+ with similar affinities, and that K1 and K2 are in the same range for all models, implying that the structuration of type B peptides is neither beneficial nor detrimental to silver binding in comparison to ultrashort HX2M and MX2H peptides. Indeed, the range of affinities is consistent with the previous hypothesis that SilE could buffer silver ions in case of high Ag+ overload, avoiding the saturation of the periplasmic adaptor SilB, in charge of the Ag+ externalization.13 Moreover, while the interaction between Ag+ ions and the sensor kinase SilS has not been investigated to date, qualitative and quantitative data are available for its homologue CusS. This homodimer protein possesses two different silver binding sites, of which one is more important for function, and is conserved between CusS and SilS.18 The complexation of the four Ag+ ions by CusS, which is also mainly carried out by His and Met residues, is governed by an apparent Kd in the μM range.19 Therefore, we propose that SilE could regulate the free Ag+ periplasmic concentration at a level to which enough Ag+ ions remain available to continuously derepress the expression of the silCFBAGP operon via SilRS, but not enough to overload SilB.
In conclusion, by means of model peptides, we qualitatively and quantitatively characterized the interaction between the different binding sequences of SilE and Ag+ ions. This study provides the first solution structures of the different silver centers found in SilE. When compared to other components of the Sil system, the characterization of the strength of Ag+/SilE interactions supports the hypothesis that SilE buffers Ag+ ions in the μM range, and hence, sustains the Ag+ export and the silCFBAGP operon expression. In view of the different metal centers in the Sil and Cus systems, it is very likely that bacteria control the metal ion transfer between the different partners of the efflux systems with the number of histidine and methionine residues involved in metal coordination.
Conflicts of interest
There are no conflicts to declare.Notes and references
- H. J. Klasen, Burns, 2000, 26, 117 CrossRef PubMed.
- J. Liu, D. A. Sonshine, S. Shervani and R. H. Hurt, ACS Nano, 2010, 4, 6903 CrossRef PubMed.
- K. Bridges, A. Kidson, E. J. Lowbury and M. D. Wilkins, Br. Med. J., 1979, 1, 446 CrossRef PubMed.
- S. L. Percival, P. G. Bowler and D. Russell, J. Hosp. Infect., 2005, 60, 1 CrossRef PubMed.
- S. Silver and L. T. Phung, Annu. Rev. Microbiol., 1996, 50, 753 CrossRef PubMed.
- S. Franke, G. Grass, C. Rensing and D. H. Nies, J. Bacteriol., 2003, 185, 3804 CrossRef PubMed.
- C. P. Randall, A. Gupta, N. Jackson, D. Busse and A. J. O’Neill, J. Antimicrob. Chemother., 2015, 70, 1037 Search PubMed.
- A. Gupta, K. Matsui, J.-F. Lo and S. Silver, Nat. Med., 1999, 5, 183 CrossRef PubMed.
- E. Elkrewi, C. P. Randall, N. Ooi, J. L. Cottell and A. J. O’Neill, J. Antimicrob. Chemother., 2017, 72, 3043 CrossRef PubMed.
- T. Vogwill and R. C. MacLean, Evol. Appl., 2015, 8, 284 CrossRef PubMed.
- S. Suzuki, T. Horinouchi and C. Furusawa, Mol. BioSyst., 2016, 12, 414 RSC.
- S. Silver, FEMS Microbiol. Rev., 2003, 27, 341 CrossRef PubMed.
- V. Chabert, M. Hologne, O. Sénèque, A. Crochet, O. Walker and K. M. Fromm, Chem. Commun., 2017, 53, 6105 RSC.
- M. Zimmermann, S. R. Udagedara, C. M. Sze, T. M. Ryan, G. J. Howlett, Z. Xiao and A. G. Wedd, J. Inorg. Biochem., 2012, 115, 186 CrossRef PubMed.
- N. Sreerama, S. Y. U. Venyaminov and R. W. Woody, Protein Sci., 2008, 8, 370 CrossRef PubMed.
- P. Kuzmič, Anal. Biochem., 1996, 237, 260 CrossRef PubMed.
- K. R. Asiani, H. Williams, L. Bird, M. Jenner, M. S. Searle, J. L. Hobman, D. J. Scott and P. Soultanas, Mol. Microbiol., 2016, 101, 731 CrossRef PubMed.
- T. Affandi, A. V. Issaian and M. M. McEvoy, Biochemistry, 2016, 55, 5296 CrossRef PubMed.
- S. A. Gudipaty and M. M. Mcevoy, Biochim. Biophys. Acta, 2014, 1844, 1656 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, binding constant determination, NMR data, structure refinement, CD spectra. See DOI: 10.1039/c8cc03784a |
| This journal is © The Royal Society of Chemistry 2018 |