
Piers’ borane-mediated hydrosilylation of epoxides and cyclic ethers†
Jianbo
Zhang
 ab,
Sehoon
Park
*ab and
Sukbok
Chang
*ab
ab,
Sehoon
Park
*ab and
Sukbok
Chang
*ab
aCenter for Catalytic Hydrocarbon Functionalizations, Institute for Basic Science (IBS), Daejeon 305-701, South Korea. E-mail: sehoonp@kaist.ac.kr; sbchang@kaist.ac.kr
bDepartment of Chemistry, Korea Advanced Institute of Science & Technology (KAIST), Daejeon 305-701, South Korea
First published on 7th June 2018
Abstract
We report the first diarylborane-catalysed hydrosilylation of epoxides and cyclic ethers. Mechanistic studies on the in situ generated Piers’ borane (C6F5)2BH with hydrosilanes in the presence of an epoxide revealed that an alkyloxy(diaryl)borane (C6F5)2BOR is readily formed as a catalytically competent species for the outer-sphere hydrosilylation of epoxides and cyclic ethers.
Epoxides are a highly useful synthetic building unit frequently employed for the construction of multi-functionalized and/or complex molecules in organic synthesis1 and polymer chemistry.2 Among various transformations, selective reduction of unsymmetrical epoxides has drawn special attention since it could selectively afford one of the two isomeric alcohol products. For instance, heterogeneous hydrogenolysis of epoxides by a Pd-based catalyst system has been well studied.3,4 Although this procedure offers a straightforward synthetic route to alcohols from epoxides, it often suffers from low selectivity and a narrow substrate scope.5 In this regard, the hydrosilylation of epoxides using well-defined homogeneous catalysts could be a competent alternative to the hydrogenolysis. In fact, a number of homogeneous catalysts have been developed for the epoxide hydrosilylation by several research groups (Scheme 1a).6 The working mode of these catalysts can be divided into four types: (i) a silylium ion-mediated outer-sphere pathway; (ii) an inner-sphere path involving an epoxide C–O bond insertion into a metal hydride; (iii) a radical process involving a metal-centered radical species; and (iv) a route via a base-initiated outer-sphere hydride transfer.
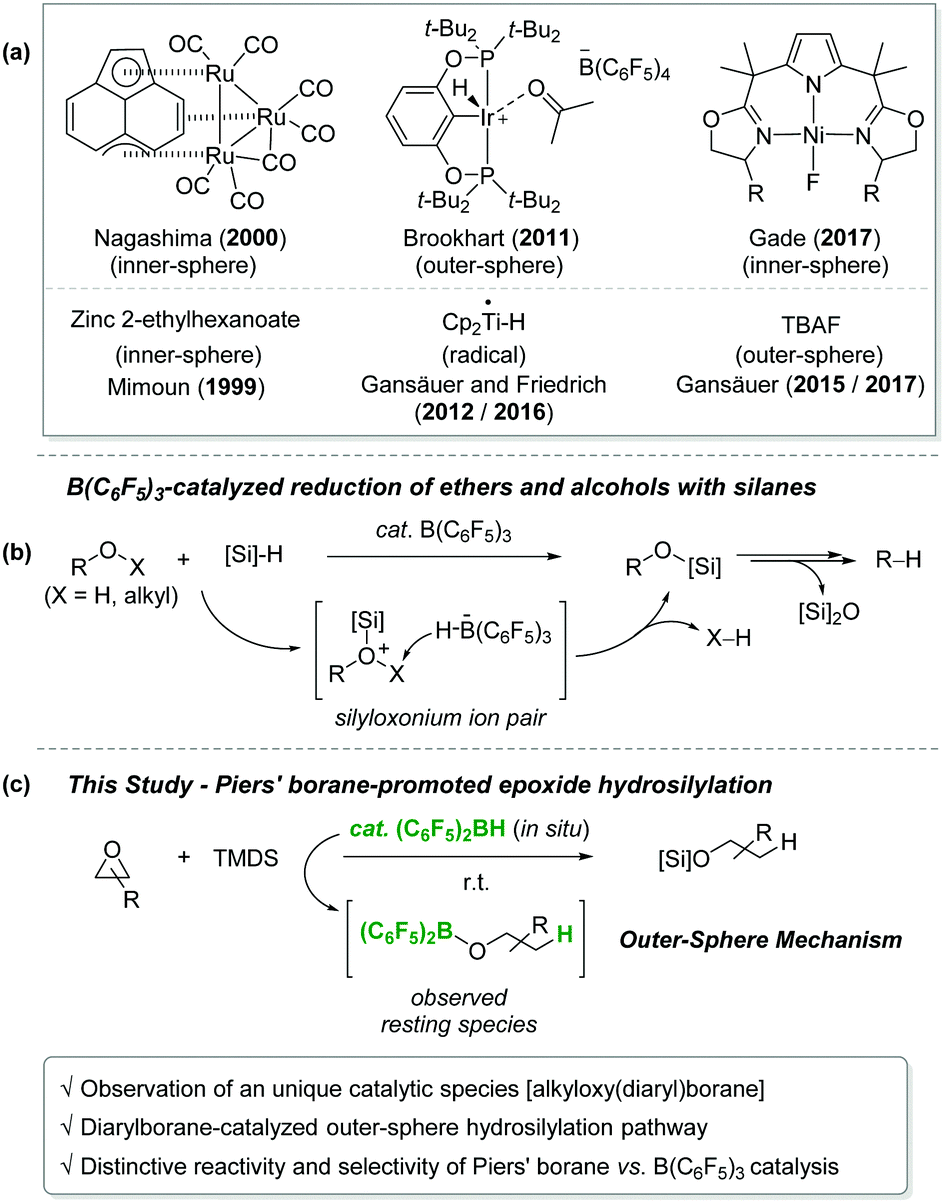 | ||
| Scheme 1 (a) Homogeneous catalysts for epoxide hydrosilylation. (b) B(C6F5)3-catalysed C–O bond cleavage of alkylethers with hydrosilanes. (c) Bis(pentafluorophenyl)borane-promoted hydrosilylation of epoxides (this work). TBAF = tetrabutylammonium fluoride, TMDS = 1,1,3,3-tetramethyldisiloxane. | ||
On the other hand, a highly electron-deficient arylborane B(C6F5)3 is known to be an efficient catalyst for the conversion of ethers and alcohols with hydrosilanes to provide a range of silyl ethers.7 One critical limitation in this procedure is an exhaustive reduction giving rise to alkanes. Such a deoxygenative path is mainly driven by intrinsically high Lewis acidity of B(C6F5)3 (Scheme 1b).7c–f The B(C6F5)3-catalysed hydrosilylative transformation has been postulated to proceed via a silyloxonium ion bearing a borohydride anion [HB(C6F5)3−], where the borohydride attacks the α-carbon of oxonium leading to the C–O bond cleavage. In this context, we hypothesized that a less Lewis acidic Piers’ borane (C6F5)2BH that is readily generated in situ from the reaction of (C6F5)2BOH with hydrosilanes can mediate the hydrosilylation of epoxides and cyclic ethers without the exhaustive deoxygenation.
Here, we report the hydrosilylation of epoxides mediated by in situ generated Piers’ borane (C6F5)2BH with an emphasis on the catalytic pathway (Scheme 1c).8 Mechanistic investigations revealed that an alkyloxy(diaryl)borane (C6F5)2BOR is formed upon the reaction of in situ generated Piers’ borane with epoxides, and that it acts as a competent catalyst for the outer-sphere hydrosilylation of epoxides. Stoichiometric studies suggested that the generation of Piers’ borane from the alkyloxyborane is slower relative to the alkyloxyborane-mediated hydrosilylation process. Most significantly, it was found that the selectivity for the ring-opening of epoxides is reversed between the Piers’ borane and the B(C6F5)3 catalyst.
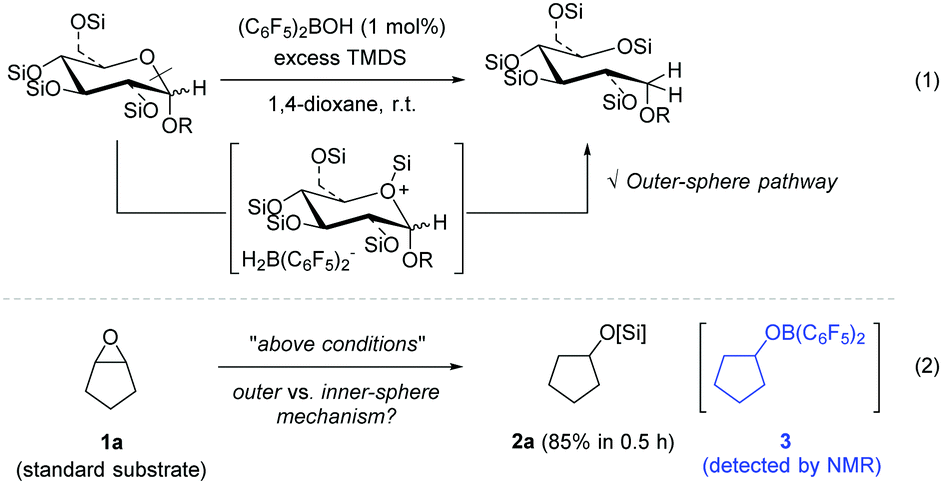
Previously, we reported a selective C–O bond cleavage of sugars via hydrosilylation catalysed by Piers’ borane (C6F5)2BH generated in situ [eqn (1)].9 This reductive transformation of sugars was proposed to proceed via an outer-sphere ionic pathway involving a cyclic silyloxonium ion bearing a borohydride [H2B(C6F5)2−], selectively providing a range of linear polyols. Based on this precedent, we were encouraged to apply the procedure for the hydrosilylation of cyclopentene oxide 1a, which was chosen as a representative substrate for preliminary mechanistic studies in an effort to elucidate the reaction pathway. As envisaged, the reaction of 1a with 1,1,3,3-tetramethyldisiloxane (TMDS) took place in the presence of (C6F5)2BOH (1 mol%) to furnish the corresponding cyclopentyloxysilane 2a in 85% yield in 0.5 h [eqn (2)]. Interestingly, 19F NMR spectroscopy of the reaction mixture exhibited a set of major signals due to cyclopentyloxy[bis(pentafluorophenyl)]borane 310 at δ −133.5, −150.9, and −162.8, in addition to a dioxane adduct with Piers’ borane, (C6F5)2BH-dioxane, as a minor species (see details in the ESI†).
To shed light on the plausible working mode, a series of catalytic and stoichiometric reactions were conducted (Scheme 2). A hydrosilylation reaction of cyclopentene oxide (1a) using TMDS-d2 as a reductant gave cyclopentyloxysilane 2a-d1 in 85% yield in 0.5 h at room temperature (Scheme 2a). This product was found to contain a deuterium incorporated exclusively at the β-position relative to the oxygen atom of the product. On the other hand, a stoichiometric treatment of cyclopentene oxide with (C6F5)2BH–SMe2 in the absence of hydrosilane afforded cyclopentyloxyborane 3 in 47% yield in 10 min at room temperature,11 whereas a reaction of cyclopentyloxyborane 3 with excess TMDS (50 equiv.) led to the formation of Piers’ borane at a relatively slower rate (10% in 0.5 h) (Scheme 2b and c).
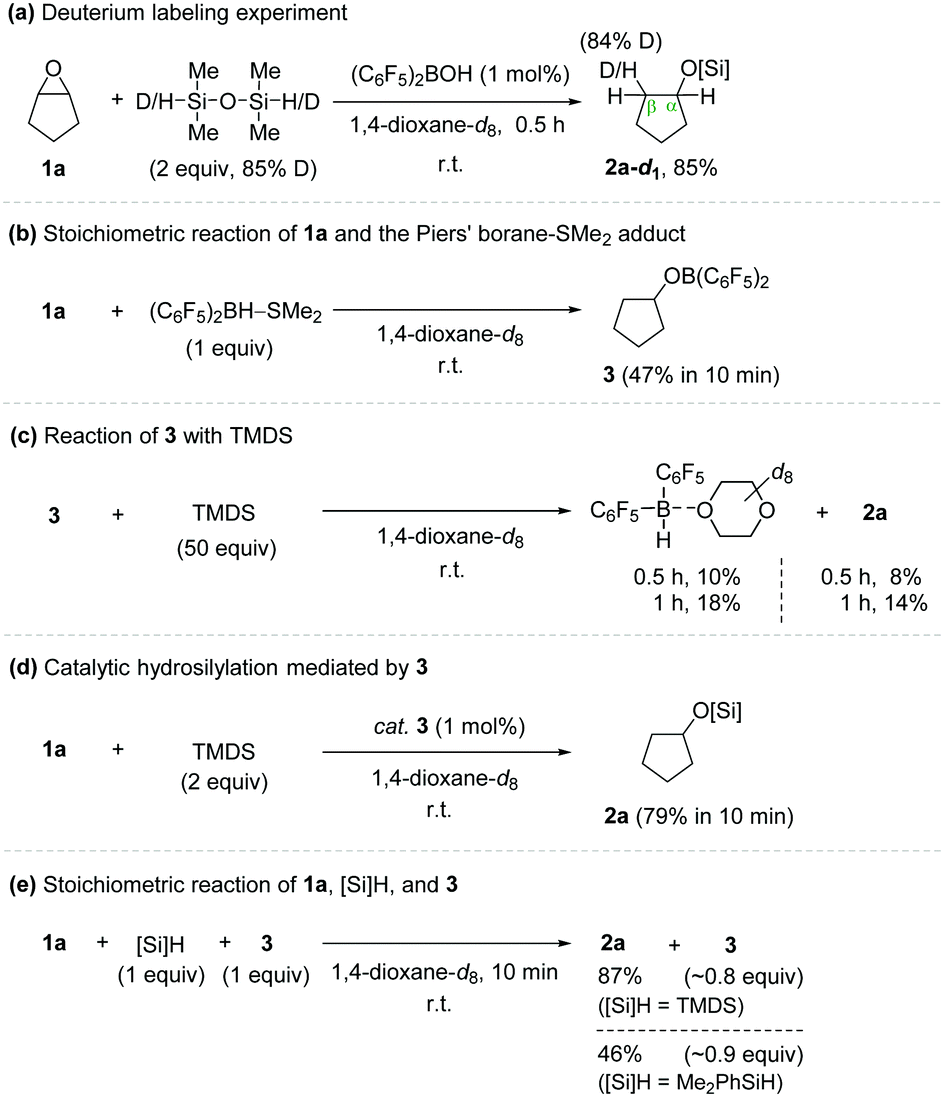 | ||
| Scheme 2 Preliminary mechanistic experiments. | ||
Notably, cyclopentyloxyborane 3 was shown to catalyse the hydrosilylation of epoxide 1a by using TMDS to furnish 2a in 79% yield in 10 min, implying that an outer-sphere ionic path is operative in this process (Scheme 2d). To obtain additional insights, a stoichiometric reaction of 1a, hydrosilanes, and 3 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1) was performed in 1,4-dioxane (Scheme 2e). Cyclopentene oxide 1a was gradually converted to 2a, and its progress was found to be dependent on the hydrosilanes employed (87% with TMDS; 46% with Me2PhSiH in 10 min). 1H and 19F NMR spectroscopy of the reaction mixtures displayed a set of major signals for 3 and minor signals for (C6F5)2BH–dioxane (see details in the ESI†).
:1:1) was performed in 1,4-dioxane (Scheme 2e). Cyclopentene oxide 1a was gradually converted to 2a, and its progress was found to be dependent on the hydrosilanes employed (87% with TMDS; 46% with Me2PhSiH in 10 min). 1H and 19F NMR spectroscopy of the reaction mixtures displayed a set of major signals for 3 and minor signals for (C6F5)2BH–dioxane (see details in the ESI†).
Based on the observation that the isolated alkoxy(bisaryl)borane (3) efficiently mediates both catalytic and stoichiometric hydrosilylation of epoxide 1a to give 2a and that the conversion of 3 to (C6F5)2BH with TMDS is rather slow, the species 3 generated in situ under the employed catalytic conditions is proposed to be a competent catalyst for the present outer-sphere ionic hydrosilylation involving a silylium ion transfer.12
Given the above experimental results, a catalytic cycle of the borane-mediated hydrosilylation of cyclopentene oxide (1a) is depicted in Scheme 3. Initially, the Piers’ borane (C6F5)2BH is assumed to be generated upon the reaction of (C6F5)2BOH with TMDS in dioxane. The in situ generated Piers’ borane would be in equilibrium with its dioxane adduct I. An epoxide substrate coordinates to the boron center of (C6F5)2BH to form an epoxide adduct II, which induces a hydroborative ring-opening of the epoxide13 to afford an alkoxy(bisaryl)borane 3. The species 3 is proposed to catalyse the outer-sphere hydrosilylation of the epoxide via a silyloxonium ion intermediacy (III), where a nucleophilic hydride transfer is highly facile to occur, releasing an O-silyl ether product 2a. An intuitive path proceeding via a direct release of product 2a from 3 with the regeneration of Piers’ borane is assumed to be kinetically less favoured.14
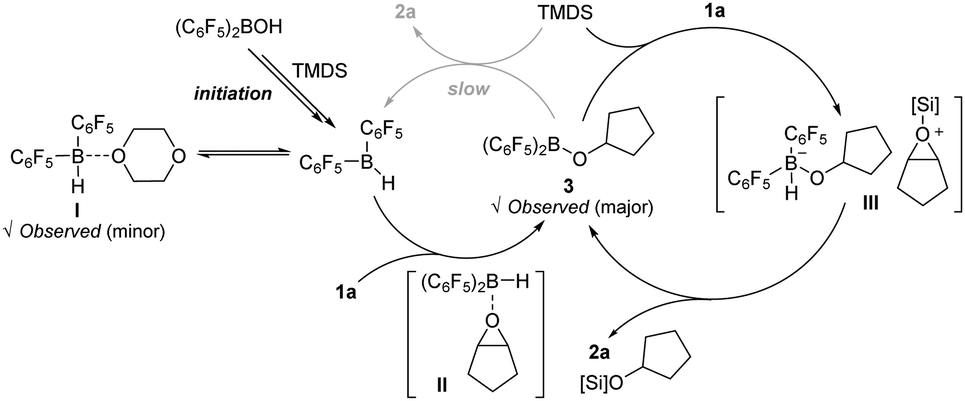 | ||
| Scheme 3 Proposed reaction pathway. | ||
Next, our proposal involving an alkoxy(bisaryl)borane as a catalytically active species (Scheme 3) led us to investigate comparative catalytic reactivity between Piers’ borane and B(C6F5)3 with regard to hydrosilylation of certain epoxides (Scheme 4a). When 2,2,3,3-tetramethyloxirane (1b) was applied as a substrate, two isomeric products were obtained depending on the borane catalysts used. With Piers’ borane generated in situ (TMDS), 2,3-dimethylbutan-2-silyl ether (2b) was formed in 97% yield, while 3,3-dimethylbutan-2-silyl ether (2b′) was obtained exclusively by using the B(C6F5)3 catalyst in combination with Et3SiH.15 This isomeric product (2b′) is assumed to be formed via a migratory ring-opening process. Although there is no compelling evidence to account for the selectivity reversal between the two borane catalysts applied, this outcome can be rationalized by the difference in hydride donor ability between the individually presupposed borohydride species, HB(C6F5)2(X)− (X = alkoxy and H) and HB(C6F5)3− (e.g. ΔΔGH− = ca. 15 kcal mol−1, when X = OPh).16 For example, a silyloxonium intermediate formed upon a silylium ion transfer undergoes a nucleophilic attack by HB(C6F5)2(X)− (X = alkoxy and H) prior to a methyl migration, whereas the same intermediate is first ring-opened and a methyl group is migrated before hydride reduction by the relatively less hydridic borohydride species HB(C6F5)3− (Scheme 4b). A similar trend of selectivity was also observed in the hydrosilylation of cis-stilbene oxide mediated by the same borane (pre)catalysts.17
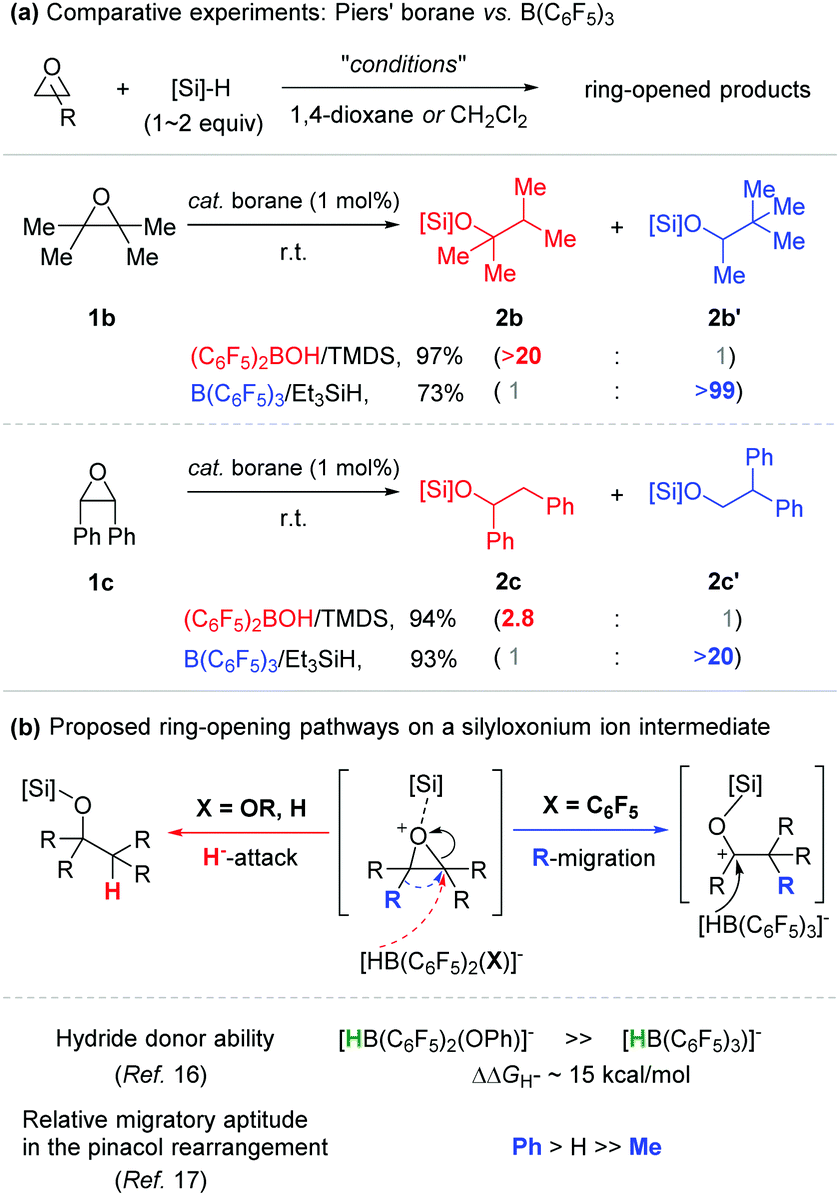 | ||
| Scheme 4 (a) Comparative experiments using in situ generated Piers’ borane vs. B(C6F5)3 (pre)catalysts. (b) Plausible pathways of epoxide ring-opening on the presupposed silyloxonium ion intermediate. | ||
Finally, the substrate scope was investigated (Scheme 5).18 A range of mono- and di-substituted epoxides and cyclic ethers was efficiently hydrosilylated at room temperature in the presence of catalytic (C6F5)2BOH (0.01–1.0 mol%). The present system proved to be compatible with functional groups such as halides, nitro, alkenyl, or alkynyl, while the product yield of an ester-containing epoxide was slightly lower. Significantly, the present system was readily amenable to gram-scale reactions: styrene oxide and tetrahydrofuran (in neat) were transformed to the corresponding products in 71% (1.74 g) and 90% (3.85 g, 9000 TON) yields, respectively (see details in the ESI†). When a chiral epoxide (98% ee) was subjected to the present conditions, an enantioenriched alcohol product was obtained (>98% ee).
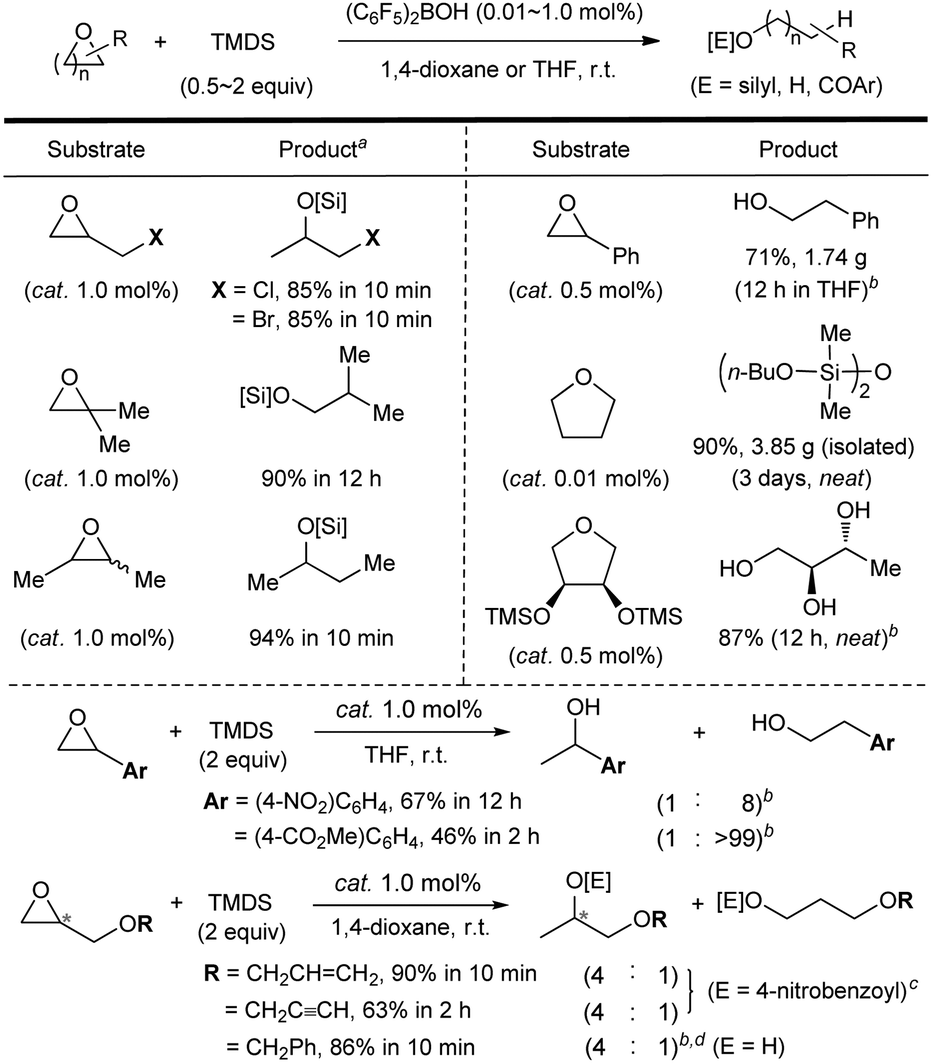 | ||
| Scheme 5 Substrate scope of the Piers’ borane-catalysed hydrosilylation. aYields were determined using 1H NMR. bThe desired alcohol products were isolated upon hydrolysis with saturated K2CO3 in MeOH. cThe crude products were O-benzoylated in situ through the reaction with 4-nitrobenzoyl chloride. d(S)-1-(Benzyloxy)propan-2-ol was obtained as a major product with >98% ee. | ||
In summary, for the first time, we have developed the Piers’ borane-catalysed hydrosilylation of epoxides and cyclic ethers. Mechanistic studies indicated that an alkyloxy(diaryl)borane is a competent catalytic species, while the reaction proceeds via an outer-sphere ionic pathway. Significantly, a selectivity reversal between Piers’ borane and B(C6F5)3 catalyst systems was observed, which could be in turn rationalized by the difference in hydride donor ability of the presupposed borohydride species. The present catalyst system is convenient to perform under mild conditions and compatible with functional groups, thus enabling applications in synthetic organic chemistry plausible.
This research was supported by the Institute for Basic Science (IBS-R010-D1), South Korea.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) S. Winstein and R. B. Henderson, in Heterocyclic Compounds, ed. R. C. Elderfield, John Wiley & Sons, New York, 1950, vol. 1, pp. 1–60 Search PubMed; (b) P. Crotti and M. Pineschi, in Aziridines and Epoxides in Organic Synthesis, ed. A. K. Yudin, Wiley-VCH, Weinheim, 2006, pp. 271–398 Search PubMed; (c) J. G. Smith, Synthesis, 1984, 629 CrossRef; (d) J. He, J. Ling and P. Chiu, Chem. Rev., 2014, 114, 8037 CrossRef PubMed.
- (a) Polymer Science: A Comprehensive Reference, ed. S. Penczek and R. H. Grubbs, Elsevier, Amsterdam, 2012, vol. 4 Search PubMed; (b) M. I. Childers, J. M. Longo, N. J. Van Zee, A. M. LaPointe and G. W. Coates, Chem. Rev., 2014, 114, 8129 CrossRef PubMed; (c) J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170 CrossRef PubMed.
- (a) P. S. Dragovich, T. J. Prins and R. Zhou, J. Org. Chem., 1995, 60, 4922 CrossRef; (b) S. V. Ley, C. Mitchell, D. Pears, C. Ramarao, J.-Q. Yu and W. Zhou, Org. Lett., 2003, 5, 4665 CrossRef PubMed; (c) E. Thiery, J. Le Bras and J. Muzart, Green Chem., 2007, 9, 326 RSC; (d) M. S. Kwon, I. S. Park, J. S. Jang, J. S. Lee and J. Park, Org. Lett., 2007, 9, 3417 CrossRef PubMed.
- Lemaire applied a catalyst system composed of Pd/C and 1,1,3,3-tetramethyldisiloxane (TMDS) for the hydrosilylation of epoxides and cyclic ethers: L. Pehlivan, E. Métay, O. Boyron, P. Demonchaux, G. Mignani and M. Lemaire, Eur. J. Org. Chem., 2011, 4687 CrossRef.
- (a) S. Mitsui, S. Imaizumi, M. Hisashige and Y. Sugi, Tetrahedron, 1973, 29, 4093 CrossRef; (b) G. C. Accrombessi, P. Geneste, J.-L. Olivé and A. A. Pavia, J. Org. Chem., 1980, 45, 4139 CrossRef; (c) H. Sajiki, K. Hattori and K. Hirota, Chem. Commun., 1999, 1041 RSC.
- (a) H. Mimoun, J. Org. Chem., 1999, 64, 2582 CrossRef; (b) H. Nagashima, A. Suzuki, T. Iura, K. Ryu and K. Matsubara, Organometallics, 2000, 19, 3579 CrossRef; (c) S. Park and M. Brookhart, Chem. Commun., 2011, 47, 3643 RSC; (d) J. Wenz, H. Wadepohl and L. H. Gade, Chem. Commun., 2017, 53, 4308 RSC; (e) A. Gansäuer, M. Klatte, G. M. Brändle and J. Friedrich, Angew. Chem., Int. Ed., 2012, 51, 8891 CrossRef PubMed; (f) D. S. G. Henriques, K. Zimmer, S. Klare, A. Meyer, E. Rojo-Wiechel, M. Bauer, R. Sure, S. Grimme, O. Schiemann, R. A. Flowers and A. Gansäuer, Angew. Chem., Int. Ed., 2016, 55, 7671 CrossRef PubMed; (g) Y.-Q. Zhang, N. Funken, P. Winterscheid and A. Gansäuer, Angew. Chem., Int. Ed., 2015, 54, 6931 CrossRef PubMed; (h) Y.-Q. Zhang, C. Poppel, A. Panfilova, F. Bohle, S. Grimme and A. Gansäuer, Angew. Chem., Int. Ed., 2017, 56, 9719 CrossRef PubMed.
- (a) J. M. Blackwell, K. L. Foster, V. H. Beck and W. E. Piers, J. Org. Chem., 1999, 64, 4887 CrossRef PubMed; (b) V. Gevorgyan, M. Rubin, S. Benson, J.-X. Liu and Y. Yamamoto, J. Org. Chem., 2000, 65, 6179 CrossRef PubMed; (c) V. Gevorgyan, M. Rubin, J.-X. Liu and Y. Yamamoto, J. Org. Chem., 2001, 66, 1672 CrossRef PubMed; (d) L. L. Adduci, M. P. McLaughlin, T. A. Bender, J. J. Becker and M. R. Gagné, Angew. Chem., Int. Ed., 2014, 53, 1646 CrossRef PubMed; (e) M. Tan and Y. Zhang, Tetrahedron Lett., 2009, 50, 4912 CrossRef; (f) C. K. Hazra, J. Jeong, H. Kim, M.-H. Baik, S. Park and S. Chang, Angew. Chem., Int. Ed., 2018, 57, 2692 CrossRef PubMed.
- Recently, Morandi reported a single example of hydrosilylation of an epoxyalcohol catalysed by B(C6F5)3: N. Drosos, G.-J. Cheng, E. Ozkal, B. Cacherat, W. Thiel and B. Morandi, Angew. Chem., Int. Ed., 2017, 56, 13377 CrossRef PubMed.
- J. Zhang, S. Park and S. Chang, Angew. Chem., Int. Ed., 2017, 56, 13757 CrossRef PubMed.
- The observed 19F NMR shifts of the cyclopentyloxyborane 3 were well matched with those of the independently synthesized borane compound. For the synthesis of alkyloxy(diaryl)boranes: (a) D. Donghi, D. Maggioni, T. Beringhelli, G. D’Alfonso, P. Mercandelli and A. Sironi, Eur. J. Inorg. Chem., 2008, 1645 CrossRef; (b) L. E. Longobardi, C. Tang and D. W. Stephan, Dalton Trans., 2014, 43, 15723 RSC.
- Although the reaction led to a quantitative conversion of 1a, the reaction mixture contained intractable ring-opened side products in addition to the alkyloxyborane 3. These side products are presumed to be formed upon a nucleophilic attack by SMe2. See details in the ESI†.
- For selected literature for outer-sphere ionic hydrosilylation: (a) M. Iglesias, F. J. Fernández-Alvarez and L. A. Oro, ChemCatChem, 2014, 6, 2486 CrossRef; (b) M. C. Lipke, A. L. Liberman-Martic and T. D. Tilley, Angew. Chem., Int. Ed., 2017, 56, 2260 CrossRef PubMed; (c) S. Park and S. Chang, Angew. Chem., Int. Ed., 2017, 56, 7720 CrossRef PubMed; (d) M. Oestreich, J. Hermeke and J. Mohr, Chem. Rev. Soc., 2015, 44, 2202 RSC; (e) N. Gandhamsetty, S. Joung, S.-W. Park, S. Park and S. Chang, J. Am. Chem. Soc., 2014, 136, 16780 CrossRef PubMed.
- (a) H. C. Brown and B. C. S. Rao, J. Am. Chem. Soc., 1960, 82, 681 CrossRef; (b) H. C. Brown and N. M. Yoon, J. Am. Chem. Soc., 1968, 90, 2686 CrossRef; (c) D. J. Pasto, C. C. Cumbo and J. Hickman, J. Am. Chem. Soc., 1966, 88, 2201 CrossRef; (d) D. J. Parks, R. E. von, H. Spence and W. E. Piers, Angew. Chem., Int. Ed. Engl., 1995, 34, 809 CrossRef.
- The (C6F5)2BH-mediated outer-sphere hydrosilylation could be a competitive pathway as we previously proposed it with regard to the hydrosilylative C–O bond cleavage of sugars9.
- The hydrosilylation of 1b with TMDS in the presence of the B(C6F5)3 catalyst gave rise to exhaustively reduced alkanes simultaneously with disproportionation of TMDS.
- Z. M. Heiden and A. P. Lathem, Organometallics, 2015, 34, 1818 CrossRef.
- A phenyl migration is generally known to be much faster than that of a methyl group in the acid-mediated pinacol rearrangement. This precedent could account for the differed product selectivity observed in the reactions of 2,2,3,3-tetramethyloxirane (>20:1) and cis-stilbene oxide (2.8:1):
(a) H. O. House and E. J. Grubbs, J. Am. Chem. Soc., 1959, 81, 4733 CrossRef;
(b) K. Nakamura and Y. Osamura, J. Am. Chem. Soc., 1993, 115, 9112 CrossRef.
- Initially formed products were a mixture of silylated compounds having several siloxane moieties of [Si], which were cleanly converted to the corresponding alcohol products upon hydrolysis.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and characterization data. See DOI: 10.1039/c8cc03741h |
| This journal is © The Royal Society of Chemistry 2018 |