
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDimensional crossover of correlated anion disorder in oxynitride perovskites†‡
Hannah
Johnston
a,
Ashley P.
Black
b,
Paula
Kayser
 a,
Judith
Oró-Solé
b,
David A.
Keen
c,
Amparo
Fuertes
*b and
J. Paul
Attfield
*a
a,
Judith
Oró-Solé
b,
David A.
Keen
c,
Amparo
Fuertes
*b and
J. Paul
Attfield
*a
aCSEC and School of Chemistry, University of Edinburgh, King's Buildings, Mayfield Road, Edinburgh, EH9 3JZ, UK. E-mail: j.p.attfield@ed.ac.uk
bInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), Campus UAB, 08193 Bellaterra, Spain
cISIS Facility, Rutherford Appleton Laboratory, Didcot, OX11 0QX, UK
First published on 1st May 2018
Abstract
A simple crossover from two-dimensional to three-dimensional correlated disorder of O and N atoms on a cubic lattice has been discovered within the Ba1−xSrxTaO2N series of perovskite oxynitrides. The crossover is driven by lattice expansion as x decreases, and provides a rapid increase in entropy due to a change from subextensive to extensive configurational entropy regimes.
Correlated disorders of atoms or magnetic moments, where local structure rules do not give rise to long range (crystallographic) order, have been identified in many crystalline materials such as water and spin ices, metal cyanides and molecular systems.1 Both two-dimensional (2D) and three-dimensional (3D) examples are known, but dimensional crossovers which are associated with changes of chemical or physical properties as new degrees of freedom become available are rare for correlated disorder. Dipolar spin-ice correlations in the pyrochlore Dy2Ti2O7 were switched from 3D to 2D behaviour through application of a magnetic field,2 but analogous atomic dimensional crossovers are not reported. Here we describe a simple strain-driven 2D to 3D crossover on a cubic lattice of correlated disorder of O and N atoms within the Ba1−xSrxTaO2N perovskites series.
AMO2N or AMON2 oxynitride perovskites have useful properties such as water-splitting photocatalysis and high dielectric constants.3,4 2D correlations of O/N atoms have been evidenced in many of these materials;5 from neutron and electron diffraction studies of SrMO2N (M = Nb, Ta),6 LaTaON2,7 and RVO2N (R = Pr, Nd)8,9 perovskites, and from electron diffraction studies of EuMO2N (M = Nb, Ta)10 and EuWON2.11 Anions within the AMO2N perovskites are locally ordered as MO4N2 octahedra where the two nitrides adopt a cis (90°) configuration which is favoured by the higher covalency of the M–N bond (Fig. 1a).12 Combining the cis configuration with the linear coordination of each nitride by two M cations results in the formation of zig zag –M–N− chains and rings that spontaneously segregate into 2D planes within the perovskite structure, as shown in Fig. 1d. This is evidenced by a partial segregation of anions within the average unit cell (Fig. 1b) that is observable by neutron diffraction due to high O/N scattering contrast. A chemical symmetry between the anion orders of AMO2N and AMON2 perovskites obtains from interchanging O and N positions so that 2D layers of cis-TaO chains are found in LaTaON2.
 | ||
Fig. 1 Anion orders in AMO2N perovskites, with N/O atoms shown as blue/red throughout. (a) Local cis (90°) coordination of N atoms in all MO4N2 octahedra. (b) The average anion distribution in 2D materials which have a tetragonal P4/mmm cell with two anion sites occupied 50% by O and 50% by N, and one site of 100% O. (c) The average anion distribution in 3D materials which have the cubic Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m perovskite structure with 67% occupancy of O and 33% N at all anion sites. (d) Local picture of cis-MN chains confined to 2D, where thick blue/thin red lines correspond to M–N–M/M–O–M connections, and the average site occupancies correspond to those in (b). (e) Local picture of cis-MN chains propagating in 3D, giving the average site occupancies shown in (c). Correlated anion disorder in AMON2 perovskites is chemically symmetric through reversal of O and N in all figures. m perovskite structure with 67% occupancy of O and 33% N at all anion sites. (d) Local picture of cis-MN chains confined to 2D, where thick blue/thin red lines correspond to M–N–M/M–O–M connections, and the average site occupancies correspond to those in (b). (e) Local picture of cis-MN chains propagating in 3D, giving the average site occupancies shown in (c). Correlated anion disorder in AMON2 perovskites is chemically symmetric through reversal of O and N in all figures. | ||
An alternative 3D distribution of the cis-chains in oxynitride perovskites is also possible (Fig. 1e), but no evidence for a 2D to 3D crossover was found in high temperature experiments. Chains were found to be confined to 2D up to the highest measured temperature of 1100 °C in neutron diffraction studies of SrTaO2N and LaTaON2.7 However, while a neutron diffraction and PDF (pair distribution function) study of BaTaO2N reported that cis-TaO4N2 octahedra were present, it was not clear whether N atoms are distributed in 2D or 3D.13 We have therefore explored the full series of solid solutions Ba1−xSrxTaO2N (0 ≤ x ≤ 1) using high resolution neutron and X-ray diffraction to establish the 2D/3D nature and evolution of the correlated anion chain disorder.
Highly crystalline powder samples of the solid solutions Ba1−xSrxTaO2N (x = 0, 0.2, 0.4, 0.6, 0.8 and 1) were prepared using stoichiometric amounts of BaCO3 (Alfa Aesar 99.997%), SrCO3 (Alfa Aesar 99.994%) and Ta3N5 (made by ammonolysis of Ta2O5 (Aldrich 99.99%) at 850 °C). A pellet was fired at 950 °C for 6 h and then 1500 °C for 3 h under a 5% H2/95% N2 gas mixture (Air Liquide). The sample was placed in a molybdenum crucible and covered with a zirconium foil to scavenge oxygen and water. Nitrogen contents from chemical analysis were found to be between 0.95(2) and 0.99(2) N atoms per formula unit without any systematic variation with Sr content x showing that the materials are essentially stoichiometric.
High resolution X-ray and neutron diffraction profiles were recorded for all of the samples at 300 K. Synchrotron X-ray powder diffraction data were measured from capillary samples at the MSPD beamline14 of the ALBA Synchrotron (Cerdanyola del Vallès, Spain). Radiation with wavelength λ = 0.6263![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Å was selected by a double Si(111) crystal monochromator and calibrated with NIST Si powder. Neutron powder diffraction data were collected on the High Resolution Powder Diffractometer (HRPD) at the ISIS spallation neutron source, Rutherford Appleton Laboratory, UK. Approximately 1 g of each sample was used in a vanadium can and diffraction patterns were recorded for 2 hours. Neutron and X-ray powder data were analysed separately by Rietveld refinement using the Fullprof program.15 In the neutron analysis, structural models were refined against data collected from the backscattering bank which provides a d range of 0.65–2.5 Å.
Å was selected by a double Si(111) crystal monochromator and calibrated with NIST Si powder. Neutron powder diffraction data were collected on the High Resolution Powder Diffractometer (HRPD) at the ISIS spallation neutron source, Rutherford Appleton Laboratory, UK. Approximately 1 g of each sample was used in a vanadium can and diffraction patterns were recorded for 2 hours. Neutron and X-ray powder data were analysed separately by Rietveld refinement using the Fullprof program.15 In the neutron analysis, structural models were refined against data collected from the backscattering bank which provides a d range of 0.65–2.5 Å.
Previous neutron studies have shown that SrTaO2N has a superstructure at 300 K due to ordered rotations of the TaO4N2 octahedra.6,7 However, the x = 1 sample used in this investigation was found to have very broad superstructure peaks indicating that the rotational domains are small, and this is confirmed by electron diffraction patterns of individual microcrystallites. Further details are in ESI.‡ Complete suppression of the superstructure was recently reported in another study where SrTaO2N was also prepared by high temperature nitrogen treatment.16 No rotational superstructure peaks were observed in the powder neutron diffraction profiles of the other Ba1−xSrxTaO2N samples. Hence fits of the simple tetragonal P4/mmm (Fig. 1b) or cubic Pmm (Fig. 1c) models were used to determine the degree of anion order for all samples.
The high Δd/d resolutions of the HRPD and MSPD instruments enable the P4/mmm model to be tested, although the tetragonal lattice distortion is extremely small and all Ba1−xSrxTaO2N patterns appear cubic by eye, as shown in Fig. 2 and ESI.‡ For x = 0.4–1.0 samples, the tetragonal P4/mmm model was found to give stable neutron refinements with consistent tetragonal strains ((a − c)/a ≈ 10−4), as shown in Fig. 3, and refined O/N site occupancies within ±7% of the ideal values for the 2D cis-chains model, as in Fig. 4. Hence the 2D correlated disorder previously found for x = 1 SrTaO2N is demonstrated to extend across the x = 0.4–1.0 range. In contrast, neutron refinements of tetragonal lattice parameters and O/N site occupancies were unstable for the x = 0 and 0.2 samples, and so these data were fitted with the cubic Pmm model that is consistent with 3D correlated disorder.
 | ||
| Fig. 2 Fit of the tetragonal P4/mmm model in Fig. 1(b) describing 2D-chains to room temperature neutron powder diffraction data for Ba0.2Sr0.8TaO2N. Residuals; χ2 = 1.17, Rwp = 8.03%. | ||
 | ||
| Fig. 3 Variations of the tetragonal strain (a − c)/a (left scale, filled points) and compressive cubic strain (a0 − a)/a0 (right scale, open points; a0 is the lattice parameter at x = 0) across the Ba1−xSrxTaO2N series with the x = 0–0.2 data from cubic perovskite structure Pmm refinements and the x = 0.4–1 data from tetragonal perovskite structure P4/mmm refinements. Red/blue points are from neutron/X-ray refinements. | ||
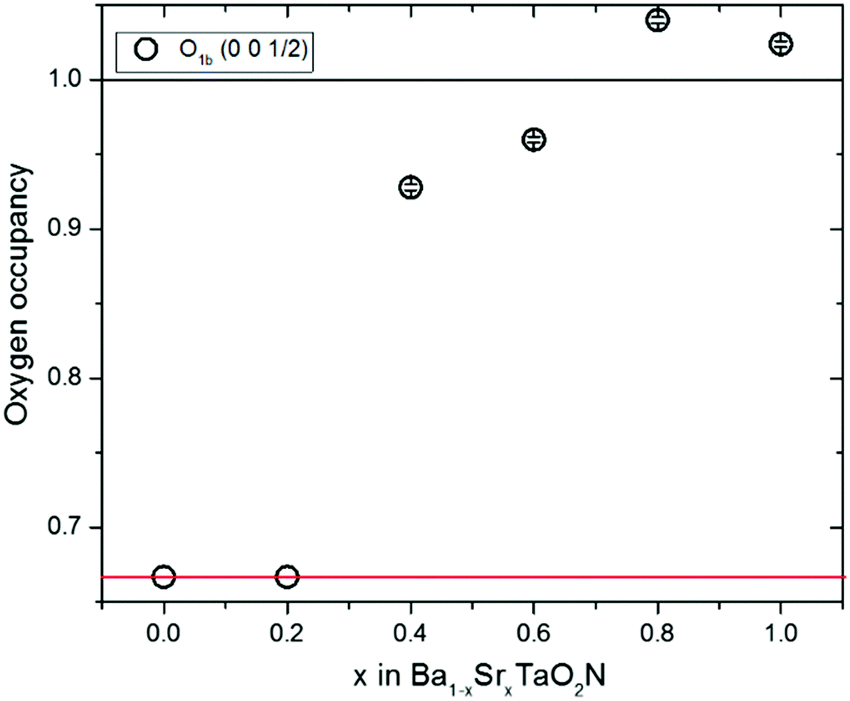 | ||
| Fig. 4 Plot of oxygen occupancies for the c-axis anion site from neutron refinements against x in the Ba1−xSrxTaO2N series. Values for the x = 0–0.2 cubic Pmm refinements are fixed at 0.67 corresponding to the model in Fig. 1c which averages over the 3D cis-TaN chains in Fig. 1e. Occupancies for the x = 0.4–1 samples are refined against the tetragonal P4/mmm model and approximate to those shown in Fig. 1b which averages the 2D cis-TaN chains model in Fig. 1d. | ||
The X-ray data do not provide O/N scattering contrast but do enable the tetragonal lattice distortion to be refined. Stable refinements of a P4/mmm tetragonal perovskite cell were obtained for x = 0.4–1.0 samples, but the refinements were unstable for fits to x = 0 and x = 0.2 data, corroborating the neutron results that these two samples have true cubic Pmm lattice symmetry. Hence both high resolution powder neutron and X-ray results demonstrate that a subtle change from cubic Pmm to tetragonal P4/mmm symmetry occurs between x = 0.2 and 0.4, with similar variations in the tetragonal lattice distortion (Fig. 3), and consistent with the neutron occupancies in Fig. 4. The variations of these quantities do not correlate with the refined temperature factors shown in ESI.‡
The cubic Ba1−xSrxTaO2N structure at x = 0–0.2 is consistent with a 3D distribution of disordered cis-chains (Fig. 1e), or at least small local domains of 2D propagation distributed over the three possible orientations, while neutron occupancies show that long range 2D confinement of anion chain layers is present across the tetragonal x = 0.4–1 samples (Fig. 1d). The absence of an intermediate P4/mmm structure with O/N occupancies between the 2D and 3D distributions (i.e. oxygen occupancy ≈70–90% in Fig. 4) indicates that the crossover is relatively sharp with different thermodynamic factors favouring 2D or 3D correlations.
Although both the 2D and 3D correlated arrangements (Fig. 1d and e) appear disordered by eye, previous theoretical analysis has shown that their configurational entropies are very different.17 The 2D arrangement found in x = 1 SrTaO2N has a subextensive molar entropy that varies with the number of atoms N per particle as S = (2Rln2)/N1/3. The highly crystalline Ba1−xSrxTaO2N powders used here have particle sizes ∼1 μm with N ≈ 1.6 × 1010 so S ≈ 6 × 10−4R and hence configurational entropy is very small for the 2D materials. This unusual structural state has been described as an ‘open order’ based on the closure properties of the set of vectors for long range structural correlation.17 The 3D arrangement of chains found in x = 0 BaTaO2N has a conventional configurational entropy (‘closed order’) that scales extensively, with value S = 2Rln(4/3) ≈ 0.6R. Hence the 2D to 3D crossover in anion chain correlation behaves thermodynamically as an order to disorder transition with an increase of 0.6R ≈ 4.8 J K−1 mol−1 in configurational entropy. This must be balanced by some ΔH = TΔS enthalpy stabilisation of the 2D state at the crossover, and as the present samples were equilibrated at 1500 °C the stabilisation of the 2D state relative to the 3D is estimated as ΔH ≈ 8.5 kJ mol−1. This stabilisation most likely derives from conjugation energy when all Ta–N bonds are co-planar, analogous to the stabilisation of planar structures for aromatic organic molecules, and so is very sensitive to increasing interatomic separation which lowers Ta–N orbital hybridisation. Hence the lattice expansion as x decreases (shown as a decreasing compressive cubic lattice strain on the right hand scale in Fig. 3) drives the 2D to 3D crossover in the Ba1−xSrxTaO2N series.
In conclusion, this study demonstrates that a sharp dimensional crossover of correlated atomic disorder occurs near x = 0.2 in the Ba1−xSrxTaO2N series. The 2D phase is destabilised by lattice expansion as x decreases, and the release of structural degrees of freedom provides a rapid increase in entropy due to a change from subextensive to extensive configurational entropy regimes.
We thank STFC, UK for support for H. J. and provision of ISIS beamtime, and EPSRC for additional support. This work was also supported by the Ministerio de Economia y Competitividad (MINECO), Spain through Project MAT2017-86616-R, the Severo Ochoa Program SEV-2015-0496, and Fellowship support to AB (MAT2011-24757). We thank the ALBA synchrotron for the provision of beamtime and Prof. Rosa Palacin, Dr Carlos Frontera (ICMAB-CSIC) and Dr F. Fauth (ALBA) for assistance with data collection.
Conflicts of interest
There are no conflicts to declare.References
- D. A. Keen and A. L. Goodwin, Nature, 2015, 521, 303 CrossRef CAS PubMed.
- Y. Tabata, H. Kadowaki, K. Matsuhira, Z. Hiroi, N. Aso, E. Ressouche and B. Fåk, Phys. Rev. Lett., 2006, 97, 257205 CrossRef CAS PubMed.
- A. Fuertes, Mater. Horiz., 2015, 2, 453 RSC.
- A. Fuertes, J. Mater. Chem., 2012, 22, 3293 RSC.
- J. P. Attfield, Cryst. Growth Des., 2013, 13, 4623 CAS.
- M. Yang, J. Oró-Solé, J. A. Rodgers, A. B. Jorge, A. Fuertes and J. P. Attfield, Nat. Chem., 2011, 3, 47 CrossRef CAS PubMed.
- L. Clark, J. Oró-Solé, K. S. Knight, A. Fuertes and J. P. Attfield, Chem. Mater., 2013, 25, 5004 CrossRef CAS.
- J. Oró-Solé, L. Clark, W. Bonin, J. P. Attfield and A. Fuertes, Chem. Commun., 2013, 49, 2430 RSC.
- L. Clark, N. Kumar, W. Bonin, A. Sundaresan, J. P. Attfield, C. N. R. Rao and A. Fuertes, J. Mater. Chem. C, 2014, 2, 2212 RSC.
- A. B. Jorge, J. Oro-Sole, A. M. Bea, N. Mufti, T. T. M. Palstra, J. A. Rodgers, J. P. Attfield and A. Fuertes, J. Am. Chem. Soc., 2008, 130, 12572 CrossRef CAS PubMed.
- M. Yang, J. Oro-Sole, A. Kusmartseva, A. Fuertes and J. P. Attfield, J. Am. Chem. Soc., 2010, 132, 4822 CrossRef CAS PubMed.
- H. Wolff and R. Dronskowski, J. Comput. Chem., 2008, 29, 2260 CrossRef CAS PubMed.
- K. Page, M. W. Stoltzfus, Y.-I. Kim, T. Proffen, P. M. Woodward, A. K. Cheetham and R. Seshadri, Chem. Mater., 2007, 19, 4037 CrossRef CAS.
- F. Fauth, I. Peral, C. Popescu and M. Knapp, Powder Diffr., 2013, 28, S360 CrossRef CAS.
- J. Rodríguez-Carvajal, Commission on Powder Diffraction (IUCr) Newsletter, 2001, 26, 12.
- D. Chen, D. Habu, Y. Masubuchi, S. Torii, T. Kamiyama and S. Kikkawa, Solid State Sci., 2016, 54, 2 CrossRef CAS.
- P. J. Camp, A. Fuertes and J. P. Attfield, J. Am. Chem. Soc., 2012, 134, 6762 CrossRef CAS PubMed.
Footnotes |
| † Open data for this paper are at https://datashare.is.ed.ac.uk/handle/10283/838. |
| ‡ Electronic supplementary information (ESI) available: N analysis, electron microscopy and further refinement results. See DOI: 10.1039/c8cc03462a |
| This journal is © The Royal Society of Chemistry 2018 |