
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Na1.5La1.5TeO6: Na+ conduction in a novel Na-rich double perovskite†
Marco
Amores
 ab,
Peter J.
Baker
c,
Edmund J.
Cussen
b and
Serena A.
Corr
*a
ab,
Peter J.
Baker
c,
Edmund J.
Cussen
b and
Serena A.
Corr
*a
aSchool of Chemistry, University of Glasgow, Glasgow G12 8QQ, UK. E-mail: Serena.Corr@glasgow.ac.uk
bDepartment of Pure and Applied Chemistry, The University of Strathclyde, Thomas Graham Building, 295 Cathedral Street, Glasgow, G1 1XL, UK
cISIS Pulsed Neutron and Muon Source, STFC Rutherford Appleton Laboratory, Harwell Science and Innovation Campus, Didcot, Oxfordshire OX11 0QX, UK
First published on 1st August 2018
Abstract
Increasing demand for lithium batteries for automotive applications, coupled with the necessity to move to large-scale energy storage systems, is driving a push towards new technologies and has seen Na-ion batteries emerge as a leading alternative to Li-ion. Amongst these, all solid-state configurations represent a promising route to achieving higher energy densities and increased safety. Remaining challenges include the need for Na+ solid electrolytes with the requisite ionic conductivities crucial for use in a solid-state cell. Here, we present the novel Na-rich double perovskite, Na1.5La1.5TeO6. The transport properties, explored at the macroscopic and local level, reveal a low activation energy barrier for Na+ diffusion and great promise for use as an electrolyte for all solid-state Na-batteries.
The lower cost and greater availability of sodium compared to lithium, together with its similar redox couple potential of ca. 0.3 V lower for Na/Na+ compared to Li/Li+, has led to enormous interest in the development of Na batteries by the energy storage community over the last decade.1 The larger cation size and heavier mass of sodium had made it a suitable candidate for medium to large-scale stationary energy storage applications, where gravimetric energy density is not a priority. As with Li-ion batteries, organic liquid Na-electrolytes present concerns in terms of safety and the operating voltage window available. The use of solid-state electrolytes in Na batteries could increase the energy density and safety of the battery, enable longer cyclability and permit the use of versatile cell geometries.2,3 Current Na+ solid-state electrolytes display Na+ conductivities at room temperature from 10−10 S cm−1, in the case of the Na(B/Al)H4 complex hydrides, to benchmark values close to the mS cm−1 for the recently reported Na-containing chalcogenides.2–4 Other systems include the NASICON phosphate materials, the classic β′′-Al2O3 and the more recent layered P2-type Na2M2TeO6 materials.2,5 The electrolyte choice should fit the specific battery application, operating temperature and electrode in order to avoid malfunction. For example, β′′-Al2O3 degrades with moisture content6 and the room temperature superionic Na-containing sulfide materials suffer from poor electrochemical stability and reaction with Na metal electrodes.7 Therefore, research on new Na+ solid-state electrolyte systems that could meet requirements lacking in present systems is critical. Here, we present a new sodium-rich solid-state electrolyte Na1.5La1.5TeO6, where the robust perovskite framework and redox stability of the Te6+ ions8–12 could enable its use in combination with high voltage electrodes.
A2BB′O6 double perovskite structure allows for a wide range of potential compositions with multiple combinations of elements on the A, B and B′ sites, where B and B′ cation positions are ordered in the crystal structure.13 The novel Na-rich Na1.5La1.5TeO6 double perovskite presented here crystallises in the monoclinic P21/n space group, with 1 mol of Na+ and 1 mol of Te6+ cations occupying the octahedral B and B′ sites in a rock-salt type ordered fashion (Fig. 1). From the 2 mols of 8-fold coordinate A-sites, 1.5 mol is occupied by La3+ cations and 0.5 mols are occupied by additional Na+, producing a “Na-rich” double perovskite with an expanded formula unit of Na0.5La1.5NaTeO6. A similar alkali-metal rich double perovskite was reported by Rosseinsky and co-workers for the Li1.5La1.5WO6 composition.14 The discrepancy between the charges and sizes of the cations sitting in the A and BB′ sites produces the distortion of the symmetry from the ideal cubic structure to a monoclinic symmetry through an a−a−b+ tilting system, as expected for a tolerance factor of 0.83 calculated for the Na1.5La1.5TeO6 composition. The synthesis of Na1.5La1.5TeO6 was achieved though an energy-efficient microwave-assisted solid-state synthesis. Such routes have been developed by our group in recent years for Li+ solid electrolytes,15–18 where the presence of hydroxide and oxide precursors increase the reaction kinetics by coupling effectively to microwave irradiation.19
 | ||
| Fig. 1 Crystallographic representation of the Na1.5La1.5TeO6 structure with monoclinic symmetry P21/n, where brown spheres represent octahedrally coordinated Te6+ ions, blue spheres represent 8-fold coordinated La3+ ions, purple spheres are the Na+ ions and the oxygen anions are represented in red. | ||
The structure of the Na1.5La1.5TeO6 double perovskite with monoclinic P21/n symmetry was established by Rietveld refinements of powder XRD data (Fig. 2). As expected, Na+ and Te6+ cations occupy the rock salt ordered B and B′ sites and refinement of site occupancies (Table S1, ESI†) indicated the presence of ∼25% vacancies on the La3+ occupying the A-site, reminiscent of the related Li1.5La1.5WO6 double perovskite.14 Na+ was refined into these vacant A-sites unoccupied by the La3+, giving an A-site occupancy for this additional Na+ of 0.239(2) which fills the available A-site vacancies. The calculated angle for the NaB–O–Te was 152.1(4)°, resulting in a Glazer tilt of 13.9°, in good agreement with the related Li1.5La1.5WO6 material (∼13.6°).14 The precision in the atomic parameters of Na+ and O2− is necessarily lower than for the stronger X-ray scatters La3+ and Te6+, but the high atomic displacement parameters of the Na+ ions may be indicative of mobility. The stoichiometry from Rietveld refinement was found to be Na1.48(1)La1.534(4)TeO6, which was further confirmed by EDX analyses, where an atomic ratio of Na1.52(8)La1.48(4)Te1.00(4) was found (Fig. S2, ESI†), in excellent agreement with XRD and the target stoichiometry. Raman analysis (Fig. S1, ESI†) also confirmed the presence of vibrational bands corresponding to the monoclinic P21/n group and the absence of carbonate or hydroxide moieties that could arise from secondary phases, typically invisible to laboratory XRD. The material microstructure was studied by SEM (Fig. 2 inset). The Na1.5La1.5TeO6 particles had sizes ranging from 1 to 5 μm, with irregular, faceted morphologies.
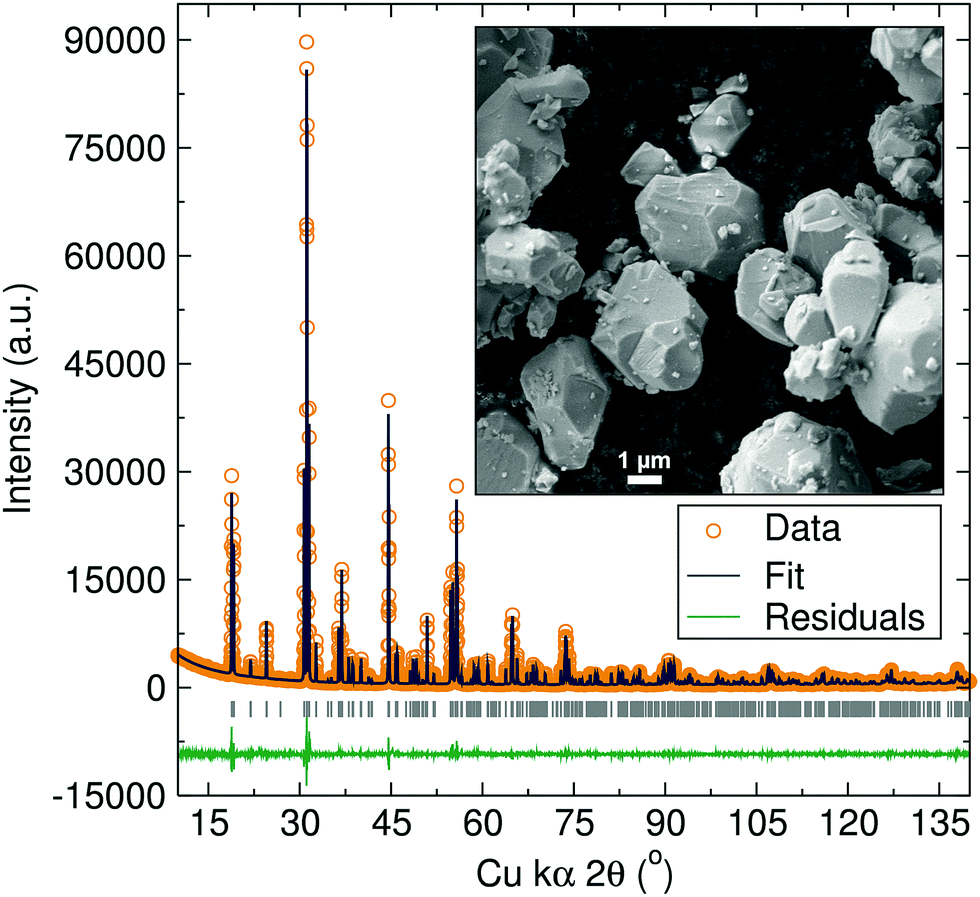 | ||
| Fig. 2 Rietveld refinements of XRD data for the Na1.5La1.5TeO6 double perovskite to the P21/n monoclinic space group. Bragg peaks positions for the monoclinic P21/n structure are indicated by vertical grey tick marks. Fit in excellent agreement to monoclinic space group P21/n, with cell parameters a = 5.69186(2) Å, b = 5.83933(2) Å, c = 8.13119(3) Å, β = 90.186(1)° and V = 270.253(1) Å3. Rwp = 0.0743, Rexp = 0.0530 and χ2 = 7.051. Inset: SEM image of Na1.5La1.5TeO6 double perovskite. | ||
To investigate the ionic macro- and microtransport properties of the Na1.5La1.5TeO6 materials, electrochemical impedance spectroscopy and muon spin relaxation measurements (μ+SR) were carried out. Fig. 3a shows the Nyquist plots of the impedance measurements, where two main components are observed. A semicircle component is noted at high frequencies, due to the resistance of the material towards ionic diffusion, and a second component in form of a linear tail can be seen at low frequencies resulting from the sodium-blocking gold electrodes employed in these measurements, indicating the predominantly ionic character of the observed impedance.20 These data were fitted using equivalent electrical circuit composed of a resistor in parallel with a constant phase element, in series with a Warburg resistance. This gave a value for the ionic conductivity of Na1.5La1.5TeO6 at room temperature of 5.4 × 10−8 S cm−1. This is on the order of that first reported for phosphate NASICON materials, which has since seen improvements up to 10−3 S cm−1 through stoichiometric and structure-tailoring strategies.21–25 These structure-tailoring strategies include element substitutions in the NASICON framework to increase the carrier concentrations or to increase the conduction channel size and hence the ionic conductivity of these materials.26,27 In the case of the P2-type Na2M2TeO6 tellurates oxides materials, the original work reported conductivities of 10−6 S cm−1 which have been now optimized to reach the mS cm−1 range.5,12 It is reasonable to expect therefore that similar improvements are possible for the Na1.5La1.5TeO6 double perovskite presented here through, for example, aliovalent doping or morphological control.
 | ||
| Fig. 3 (a) Nyquist plot of EIS data for Na1.5La1.5TeO6 at different temperatures and a representative fit to the equivalent electrical circuit. (b) Room temperature μ+SR data collected for Na1.5La1.5TeO6 at zero field and applied longitudinal fields of 5 G and 10 G, fitted (solid lines) using the Keren function. (c) Arrhenius plots of the ionic conductivity and diffusion coefficient of Na1.5La1.5TeO6 from μ+SR and EIS. | ||
The activation energy required for macroscopic ionic conduction, calculated from an Arrhenius plot of EIS data, showed a promising value of 0.27(2) eV (Fig. 3c). This value is significantly lower compared to that obtained for the analogous Li1.5La1.5WO6 double perovskite material of 0.50(5) eV.14 This lower energetic requirement for ionic diffusion found for Na1.5La1.5TeO6 could be related to the high relative density of the sintered material, nearly 96%, which results in a lower inter-grain energy requirement for Na+ diffusion between sintered particles. The macroscopic activation energy found for this Na-rich double perovskite is similar to current benchmark systems with low activation energy values from EIS measurements,2,3,28 for example Na3PSe4 which displays an activation energy of 0.21 eV.29 Preliminary EIS analysis of a Na-metal electrode-sandwiched Na1.5La1.5TeO6 cell at 80 °C revealed the lack of a Warburg element, confirming Na+ as the conducting species (Fig. S3, ESI†). An increase in impedance over time is observed, which may point to a reaction with the sodium metal electrode at this temperature. Further examination of this, together with the use of similar protection approaches used for Li-metal anodes, could overcome these compatibility issues at high temperatures.30,31
The local Na+ diffusion properties were also investigated by μ+SR measurements performed on the EMU instrument at the ISIS Neutron and Muon source. The use of μ+SR as a local probe to study Li+ and Na+ diffusion in battery materials has grown in recent years.15,32–34 The natural abundance of the spin 1/2 23Na isotope is 100%, making Na+ an ideal candidate to be studied by μ+SR. The temporal evolution of the decay positron asymmetry for Na1.5La1.5TeO6 at three different longitudinal magnetic fields is shown in Fig. 3b. At short times, the decay positron asymmetry followed a moderate decay, while at longer times the asymmetry decrease followed a slower trend, as expected for this Na-rich double perovskite with no paramagnetic ions in its structure and which also contains active nuclear spins (23Na, 139La and 125Te) which can interact with the muon spin. To obtain the fluctuation rate of the muons due to sodium-ion diffusion (Fig. S4, ESI†), the muon decay asymmetry data were fitted using Keren's analytic generalization of the Abragam function (Fig. 3b).35 The Na+ diffusion coefficients from the muon fluctuation rate at different temperatures were then calculated applying eqn (1),36 where Ni is the number of accessible Na sites in the i-th path, Zv,i is the vacancy fraction of the destination sites, si the jump distance between Na+ sites, and ν the calculated muon fluctuation rate at each temperature.
 | (1) |
Since a detailed model of the Na+ diffusion pathways for this novel Na-rich double perovskite is still not available, a simplified model based on the calculations reported by Rosseinsky and co-workers for the Li1.5La1.5WO6 perovskite was employed.14 The model is shown in Fig. S5 (ESI†) where Na+ diffuses from the A sites to the two neighbouring B sites and from the B sites to the four neighbouring A sites, creating a 3D network for Na+ diffusion. Additionally, a conservative 0.01 vacancy fraction in the Na+ positions was introduced in both A and B sites to allow diffusion. From this preliminary model, and introducing the two different NaA–NaB distances of 3.33 and 3.46 Å, a Na+ diffusion coefficient at room temperature of 4.2 × 10−12 cm2 s−1 was obtained. This diffusion coefficient is similar to that reported for the Nax(Mn/Co)O2 layered cathode materials (10−11–10−12 cm2 s−1), indicating excellent compatibility of these components in terms of Na+ diffusion.34,37,38 The microscopic diffusion of Na1.5La1.5TeO6 is also in line with other Na+ ionic conductors such as the NaxWO2Cl2 tungsten bronze with a Na+ diffusion coefficient of 10−13 cm2 s−1,39 and Na3PS4 with a value in the range of 10−12 cm2 s−1.40 The μ+SR measurements indicate that the activation energy required for local Na+ diffusion is 0.163(9) eV. This low activation energy is similar to related oxide materials, such as the Ga-doped Na2Zn2TeO6 with an activation energy of 0.12 eV as observed from NMR measurements,12 or β′-alumina single crystals with activations energies in the 0.12–0.16 eV range.41
The higher activation energy values obtained from the macroscopic EIS measurements which probes long range Na+ conduction through multiple intra-grain crystalline sites and grain boundaries, compared to microscopic μ+SR measurements which are more sensitive to individual Na+ hops within the crystalline grain, could have its origin in the contribution to the resistance from grain boundaries to ionic conduction which is virtually invisible to μ+SR. Lower conductivity values and higher energy barriers to diffusion could also result from the presence of La3+ ions in the A-sites, which could hinder long-range Na+ conductivity. Nevertheless, the excellent microscopic Na+ transport properties displayed by this novel Na1.5La1.5TeO6 Na-rich double perovskite indicates that, upon further optimization, this material could be a promising candidate as a solid electrolyte for Na batteries.
In conclusion, we have demonstrated the synthesis of a novel Na-rich double perovskite, Na1.5La1.5TeO6 through microwave methods, which crystallises with monoclinic P21/n space group with Na+ on both the A- and B-sites. The material displays a macroscopic ionic conductivity on the order of 10−8 S cm−1 at room temperature with a low activation energy of 0.27(2) eV. μ+SR measurements reveal a microscopic diffusion coefficient for Na+ at room temperature in the order of 10−12 cm2 s−1 and a very low activation energy of 0.163(9) eV. These findings reveal the promising transport properties for this Na1.5La1.5TeO6 material and further developments are forthcoming to optimize the macroscopic transport to its maximum microscopic potential.
The authors gratefully acknowledge technical support from Michael Beglan. The authors also thank the EPSRC for funding (EP/N001982/1), the STFC for beamtime allocation, the Universities of Glasgow and Strathclyde for support and the use of facilities, and the School of Chemistry at Glasgow for PhD studentship funding.
Conflicts of interest
There are no conflicts to declare.References
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC.
- J.-J. Kim, K. Yoon, I. Park and K. Kang, Small Methods, 2017, 1, 1700219 CrossRef.
- H. Che, S. Chen, Y. Xie, H. Wang, K. Amine, X.-Z. Liao and Z.-F. Ma, Energy Environ. Sci., 2017, 10, 1075–1101 RSC.
- K. B. Hueso, M. Armand and T. Rojo, Energy Environ. Sci., 2013, 6, 734–749 RSC.
- M. A. Evstigneeva, V. B. Nalbandyan, A. A. Petrenko, B. S. Medvedev and A. A. Kataev, Chem. Mater., 2011, 23, 1174–1181 CrossRef.
- F. G. Will, J. Electrochem. Soc., 1976, 123, 834–836 CrossRef.
- Z. Yu, S.-L. Shang, Y. Gao, D. Wang, X. Li, Z.-K. Liu and D. Wang, Nano Energy, 2018, 47, 325–330 CrossRef.
- A. Gupta, C. B. Mullins and J. B. Goodenough, J. Power Sources, 2013, 243, 817–821 CrossRef.
- M. Sathiya, K. Ramesha, G. Rousse, D. Foix, D. Gonbeau, K. Guruprakash, A. S. Prakash, M. L. Doublet and J.-M. Tarascon, Chem. Commun., 2013, 49, 11376–11378 RSC.
- Z. Yang, Y. Jiang, L. Deng, T. Wang, S. Chen and Y. Huang, J. Power Sources, 2017, 360, 319–323 CrossRef.
- E. A. Zvereva, V. B. Nalbandyan, M. A. Evstigneeva, H.-J. Koo, M.-H. Whangbo, A. V. Ushakov, B. S. Medvedev, L. I. Medvedeva, N. A. Gridina, G. E. Yalovega, A. V. Churikov, A. N. Vasiliev and B. Büchner, J. Solid State Chem., 2015, 225, 89–96 CrossRef.
- Y. Li, Z. Deng, J. Peng, E. Chen, Y. Yu, X. Li, J. Luo, Y. Huang, J. Zhu, C. Fang, Q. Li, J. Han and Y. Huang, Chem. – Eur. J., 2018, 24, 1057–1061 CrossRef PubMed.
- S. Vasala and M. Karppinen, Prog. Solid State Chem., 2015, 43, 1–36 CrossRef.
- A. B. Santibanez-Mendieta, C. Didier, K. K. Inglis, A. J. Corkett, M. J. Pitcher, M. Zanella, J. F. Shin, L. M. Daniels, A. Rakhmatullin, M. Li, M. S. Dyer, J. B. Claridge, F. Blanc and M. J. Rosseinsky, Chem. Mater., 2016, 28, 7833–7851 CrossRef.
- M. Amores, T. E. Ashton, P. J. Baker, E. J. Cussen and S. A. Corr, J. Mater. Chem. A, 2016, 4, 1729–1736 RSC.
- M. Amores, S. A. Corr and E. J. Cussen, J. Electrochem. Soc., 2017, 164, A6395–A6400 CrossRef.
- H. El-Shinawi, E. J. Cussen and S. A. Corr, Dalton Trans., 2017, 46, 9415–9419 RSC.
- H. El-Shinawi, G. W. Paterson, D. A. MacLaren, E. J. Cussen and S. A. Corr, J. Mater. Chem. A, 2017, 5, 319–329 RSC.
- H. J. Kitchen, S. R. Vallance, J. L. Kennedy, N. Tapia-Ruiz, L. Carassiti, A. Harrison, A. G. Whittaker, T. D. Drysdale, S. W. Kingman and D. H. Gregory, Chem. Rev., 2014, 114, 1170–1206 CrossRef PubMed.
- J. T. S. Irvine, D. C. Sinclair and A. R. West, Adv. Mater., 1990, 2, 132–138 CrossRef.
- F. E. Mouahid, M. Bettach, M. Zahir, P. Maldonado-Manso, S. Bruque, E. R. Losilla and M. A. G. Aranda, J. Mater. Chem., 2000, 10, 2748–2757 RSC.
- N. Anantharamulu, K. Koteswara Rao, G. Rambabu, B. Vijaya Kumar, V. Radha and M. Vithal, J. Mater. Sci., 2011, 46, 2821–2837 CrossRef.
- H. Park, K. Jung, M. Nezafati, C.-S. Kim and B. Kang, ACS Appl. Mater. Interfaces, 2016, 8, 27814–27824 CrossRef PubMed.
- S. Song, H. M. Duong, A. M. Korsunsky, N. Hu and L. Lu, Sci. Rep., 2016, 6, 32330 CrossRef PubMed.
- P. Colomban, Solid State Ionics, 1986, 21, 97–115 CrossRef.
- K. Arbi, J. M. Rojo and J. Sanz, J. Eur. Ceram. Soc., 2007, 27, 4215–4218 CrossRef.
- A. Martínez-Juárez, C. Pecharromán, J. E. Iglesias and J. M. Rojo, J. Phys. Chem. B, 1998, 102, 372–375 CrossRef.
- V. S. Kandagal, M. D. Bharadwaj and U. V. Waghmare, J. Mater. Chem. A, 2015, 3, 12992–12999 RSC.
- L. Zhang, K. Yang, J. Mi, L. Lu, L. Zhao, L. Wang, Y. Li and H. Zeng, Adv. Energy Mater., 2015, 5, 1501294 CrossRef.
- X.-B. Cheng, R. Zhang, C.-Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef PubMed.
- S. Wenzel, T. Leichtweiss, D. A. Weber, J. Sann, W. G. Zeier and J. Janek, ACS Appl. Mater. Interfaces, 2016, 8, 28216–28224 CrossRef PubMed.
- T. E. Ashton, J. V. Laveda, D. A. MacLaren, P. J. Baker, A. Porch, M. O. Jones and S. A. Corr, J. Mater. Chem. A, 2014, 2, 6238–6245 RSC.
- J. V. Laveda, B. Johnston, G. W. Paterson, P. J. Baker, M. G. Tucker, H. Y. Playford, K. M. Jensen, S. J. Billinge and S. A. Corr, J. Mater. Chem. A, 2018, 6, 127–137 RSC.
- M. Månsson and J. Sugiyama, Phys. Scr., 2013, 88, 068509 CrossRef.
- A. Keren, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 10039–10042 CrossRef.
- An Introduction to Solid State Diffusion, ed. R. J. Borg, G. Dienes, Academic Press, San Diego, 1988 Search PubMed.
- A. Mendiboure, C. Delmas and P. Hagenmuller, J. Solid State Chem., 1985, 57, 323–331 CrossRef.
- A. Bhide and K. Hariharan, Solid State Ionics, 2011, 192, 360–363 CrossRef.
- P. G. Bruce, J. Nowinski and V. C. Gibson, Solid State Ionics, 1992, 50, 41–45 CrossRef.
- Z. Zhu, I.-H. Chu, Z. Deng and S. P. Ong, Chem. Mater., 2015, 27, 8318–8325 CrossRef.
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc03367f |
| This journal is © The Royal Society of Chemistry 2018 |