
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantitative synthesis of protein–DNA conjugates with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h2_char_2009.gif) :1 stoichiometry†
:1 stoichiometry†
Xiaowen
Yan
,
Hongquan
Zhang
 *,
Zhixin
Wang
,
Hanyong
Peng
,
Jeffrey
Tao
,
Xing-Fang
Li
and
X.
Chris Le
*
*,
Zhixin
Wang
,
Hanyong
Peng
,
Jeffrey
Tao
,
Xing-Fang
Li
and
X.
Chris Le
*
Division of Analytical and Environmental Toxicology, Department of Laboratory Medicine and Pathology, University of Alberta, Edmonton, Alberta T6G2G3, Canada. E-mail: hongquan@ualberta.ca; xc.le@ualberta.ca
First published on 19th June 2018
Abstract
We describe here a binding-facilitated reaction strategy, enabling quantitative conjugation of DNA to native proteins with a desirable 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry. The technique takes advantage of the iterative affinity interaction and covalent binding processes to achieve complete conjugation. The complete conjugation obviates the need for separation of the protein–DNA conjugates as required by other DNA–protein conjugation methods.
:1 stoichiometry. The technique takes advantage of the iterative affinity interaction and covalent binding processes to achieve complete conjugation. The complete conjugation obviates the need for separation of the protein–DNA conjugates as required by other DNA–protein conjugation methods.
Protein–DNA conjugates, which combine the unparalleled programmability of DNA with the tremendous diversity of protein functions, have shown diverse applications, ranging from protein immobilization1–4 and bioanalysis5–9 to fluorescence imaging10–13 and targeted drug delivery.14 Several chemical conjugation methods based on either covalent bond formation or non-covalent interaction have been developed for the synthesis of protein–DNA conjugates.15–17 Among them, bi-functional cross-linkers, often present in a large excess, are most commonly used for coupling DNA to proteins through random reactions with lysine residues on the surface of proteins. Because multiple lysine residues may be present on a protein surface, such conjugation approaches result in a mixture of highly heterogeneous products with various DNA
:protein stoichiometries. Random conjugation of multiple DNA strands to a protein molecule can adversely affect the biological functions of proteins.18
Highly desirable 1:1 protein–DNA conjugation has been mainly achieved by genetically incorporating specific chemical handles into proteins of interest for subsequent DNA coupling. Several strategies using either bio-orthogonal handle,18,19 enzyme-catalyzed tag,20 or self-labeling enzyme21,22 have been developed to prepare such genetically modified proteins. However, these processes are laborious, time-consuming, and technically challenging. To achieve the 1:1 DNA–protein conjugation without the need for genetic modification of proteins, affinity molecules can be used to controllably deliver the DNA strands to proteins of interest. DNA-templated synthesis (DTS)23 has been applied to the conjugation of small molecule binding proteins with photoreactive DNA molecules.24–26 However, to date, synthesis of protein–DNA conjugates with a 1:1 stoichiometry and quantitative 100% yield has not been demonstrated. Gothelf and coworkers combined metal affinity binding and DTS for conjugation of DNA to proteins bound to metal affinity probes, such as antibodies, His-tagged proteins, and other metal-binding proteins.27,28 Their method “yielded the desired single-oligonucleotide conjugates in approximately 10% after purification by gel extraction”.27 Tan et al. used aptamer binding to achieve the conjugation of DNA to target proteins.29–31 The purified protein conjugate was “extracted from non-denaturing PAGE, and the extraction yield was about 56%”.31 Both methods required separation of protein–DNA conjugates from the unconjugated proteins. The primary objective of this research is to establish a new strategy that is able to synthesize protein–DNA conjugates with complete yield, thus obviating the need for separation or purification of the protein–DNA conjugates.
Here we describe a binding-facilitated reaction strategy enabling the quantitative synthesis of protein–DNA conjugates with a precise 1:1 stoichiometry and under mild reaction conditions (Scheme 1). This strategy makes use of affinity binding to secure the 1:1 protein–DNA conjugation and to produce high conjugation efficiency. To achieve a 1:1 protein–DNA conjugation, we introduced the 1:1 protein–ligand binding to ensure that only a single DNA strand is brought in close proximity to each protein molecule (Scheme 1a and Fig. S1a in ESI†). A DNA hybrid was first prepared by hybridizing the conjugation strand with its complementary strand; the conjugation strand was labeled with a reactive group N-hydroxysuccinimide (NHS) ester at the 5′-end and the complementary strand was labeled with an affinity ligand at the 3′-end. To avoid any random conjugation of DNA to protein, we mixed the target protein and DNA hybrid at a concentration that prevented NHS groups from directly reacting with lysine residues on the protein surfaces without the involvement of affinity binding. Because the affinity ligand allowed for the binding of a single ligand molecule to each protein molecule, only one NHS-containing DNA strand was brought in close proximity to the protein surface (Scheme S1a, ESI†), resulting in a high local concentration sufficient for covalent amide formation between NHS and the lysine residue (Scheme S1b, ESI†). As a result, 1:1 protein–DNA conjugates were formed. The binding-facilitated reaction technique was demonstrated by the successful conjugation of DNA to carbonic anhydrase II (CAII) and to α-thrombin. These reactions used a small molecule and aptamers as affinity ligands whose dissociation constants (Kd) vary by three orders of magnitude (from 0.5 nM to 3.2 μM), suggesting the general applicability.
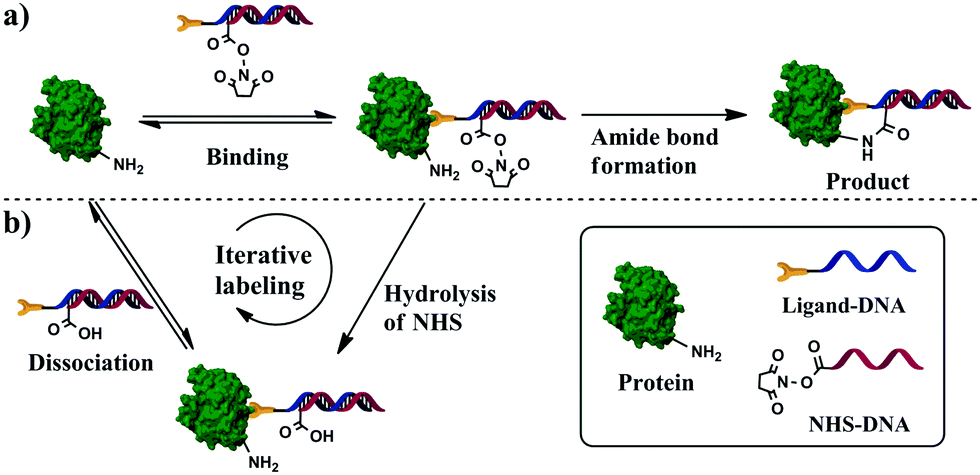 | ||
| Scheme 1 Binding-facilitated synthesis of protein–DNA conjugates using an affinity ligand and the NHS-labeled DNA strand. (a) The 1:1 protein–ligand binding brings a single NHS-DNA molecule to close proximity with the protein molecule. The NHS-DNA strand reacts with the protein by formation of a covalent amide bond between the NHS and a lysine residue. As a result, a 1:1 protein–DNA conjugate is formed. (b) NHS can also hydrolyze in aqueous solution. The DNA strand with hydrolyzed NHS cannot react with the protein. Because the affinity binding between the ligand and the protein is reversible, the DNA strand with hydrolyzed NHS can dissociate from the protein, leaving the protein available to bind with another active NHS-DNA probe. These iterative affinity interaction and covalent binding processes continue until all the protein molecules are conjugated with the DNA, in a 1:1 stoichiometry. | ||
The NHS ester can also be hydrolyzed in aqueous solutions (Scheme S1c, ESI†), which is beneficial for minimizing random protein–DNA conjugation. However, the hydrolysis of NHS esters reduces the conjugation efficiency, as in the case when NHS esters bound to DNA hybrids were degraded (Scheme S1c, ESI†). Because the affinity binding between ligands and proteins is reversible, the bound DNA hybrid containing the hydrolyzed ester can be displaced by an intact DNA hybrid, allowing unconjugated protein molecules to react with the reactive DNA hybrids (Scheme S1d, ESI†). Such iterative reaction continues until all protein molecules are conjugated.
We first used carbonic anhydrase II (CAII) and its inhibitor, benzenesulfonamide (SA), to test the feasibility of our strategy (Fig. 1a). CAII has excellent stability and well-characterized ligand-binding property.32 To prepare SA–DNA (Fig. 1b), we labeled 4-(2-aminoethyl)benzenesulfonamide (SA) with thiolated DNA using N-succinimidyl 6-maleimidocaproate (EMCS) as the cross-linker (Fig. S1a, ESI†). We used electrospray ionization mass spectrometry to monitor the reaction process and to detect the product (SA-maleimide) (Fig. S1b, ESI†). Polyacrylamide gel electrophoresis (PAGE) analysis showed that approximately 97% of DNA was labeled with SA (Fig. 1c).
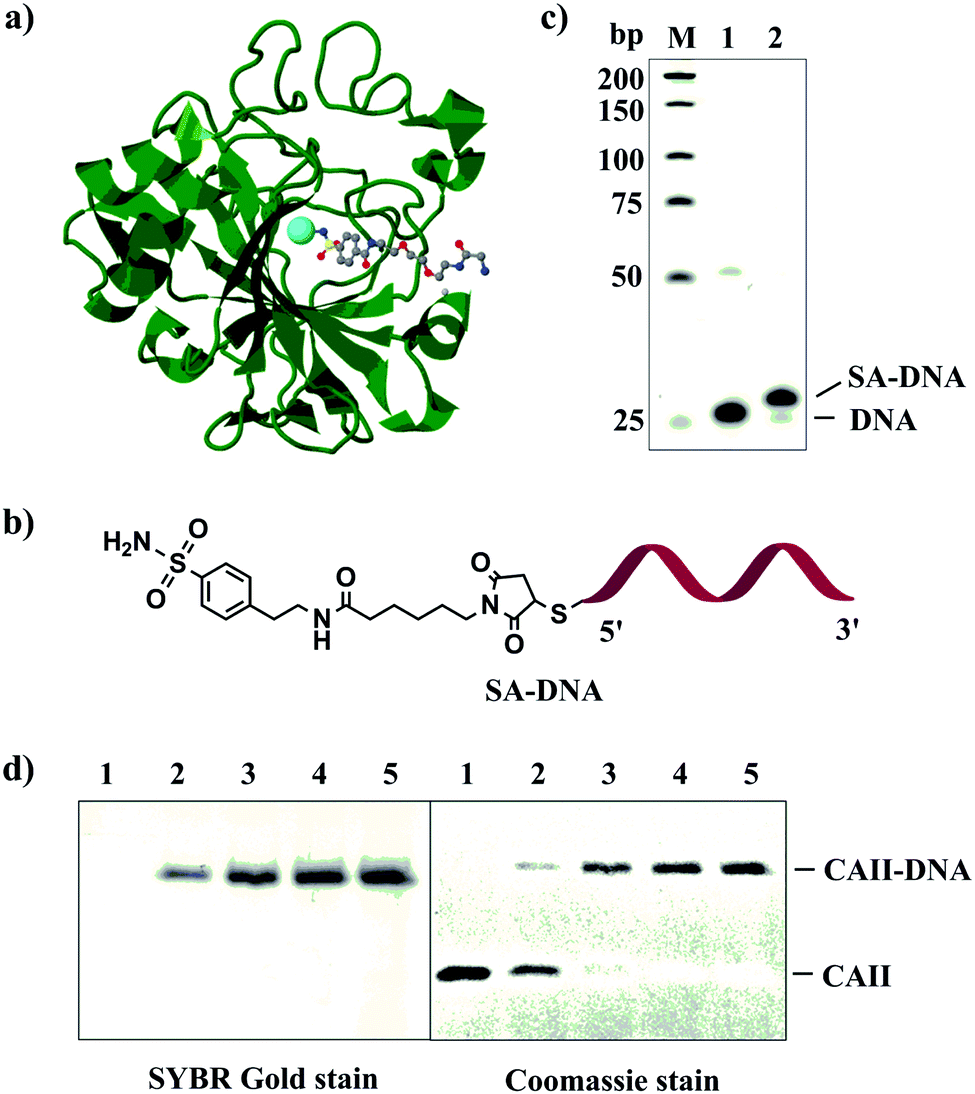 | ||
| Fig. 1 (a) Structure of bovine carbonic anhydrase II (CAII) in complex with a benzenesulfonamide compound (PDB entry 1CNW) shown in a ball & stick model. The cofactor of CAII, Zn(II), is shown as a cyan sphere. This figure was prepared using J mol: an open-source Java viewer for chemical structures in 3D. http://www.jmol.org/. (b) Structure of SA–DNA. (c) Denaturing PAGE characterizing reaction mixtures of SA–DNA. Lane M, low molecular weight DNA ladder; lane 1, thiolated DNA; lane 2, SA–DNA. (d) Reducing (2-mercaptoethanol) SDS-PAGE (12%) characterizing the conjugation products of CAII–DNA with varying DNA:protein ratios. Concentration of CAII was kept at 0.1 μM, and concentrations of SA/NHS-DNA were 0 (lane 1), 0.1 μM (lane 2), 0.2 μM (lane 3), 0.5 μM (lane 4), and 1 μM (lane 5). PAGE gels were stained with SYBR Gold and Coomassie blue to visualize protein and DNA, respectively. | ||
Given that a mild reaction condition can help to maintain the activity of the proteins, and that the amide bond formed between carbonic acid and lysine residue is very stable,27,31 we chose NHS ester as the reactive group. The NHS ester containing DNA (NHS-DNA) was prepared by reacting amine-functionalized DNA with disuccinimidyl suberate (DSS).
We have optimized the reaction conditions, such as the concentration of NHS-DNA (Fig. S2, ESI†), the reaction time (Fig. S3, ESI†), and the design of the spacer (Fig. S4, ESI†), to minimize the binding-independent background and maximize the yield (Fig. S5, ESI†). These results also indicate the importance of using affinity binding to guide the conjugation of DNA to CAII.
We used 1:1 molar ratio of DNA and CAII in the conjugation reactions. Although the conjugation of DNA to CAII was fast, the yield was not quantitative: about 62% when the concentration of CAII and the DNA was 1 μM (Fig. S4, ESI†). We hypothesized that the incomplete conjugation could be attributed to two possible reasons: poor affinity binding between CAII and SA–DNA or the hydrolysis of NHS and loss of its reactivity before NHS-DNA is brought to the surface of CAII. To test the first hypothesis, we conjugated DNA to streptavidin using biotin–DNA and NHS-DNA. We prepared the DNA hybrid between biotin–DNA and NHS-DNA and then mixed streptavidin with the hybrid at varying DNA:streptavidin ratios. SDS-PAGE analysis confirmed the conjugation yield of DNA to streptavidin was only ∼28% for the different DNA:streptavidin ratios (Fig. S6, ESI†). Because of the very high binding affinity (Kd = 10−15) of biotin to streptavidin, all streptavidin molecules were bound by the DNA hybrid, suggesting that the incomplete conjugation efficiency is not due to low binding affinity but due to the hydrolysis of NHS-DNA.
To address the issue of the hydrolysis of the NHS ester, we reasoned that the reversible association and dissociation property of the ligand–protein interaction could be used to our advantage because the dissociation of the hydrolyzed NHS-DNA would be replaced with an intact NHS-DNA hybrid. Such iterative reaction would continue until all the protein molecules are conjugated. We tested our hypothesis by increasing the DNA to CAII molar ratio from 1:1 to 2:1, 5:1 and 10:1, by increasing the SA/NHS-DNA concentration from 0.1 to 0.2, 0.5 and 1.0 μM while keeping the concentration of CAII constant at 0.1 μM. The conjugation yield increased with increasing SA/NHS-DNA concentrations. When the concentration of SA/NHS-DNA was 0.5 and 1.0 μM (lane 4 and 5, Fig. 1d), no free CAII was observed, demonstrating quantitative conjugation of DNA to CAII. These results show the first example of achieving complete synthesis of protein–DNA conjugate with a 1:1 stoichiometry.
To confirm that the cycling of association and dissociation of affinity binding leads to the complete conjugation, we conducted a competitive binding experiment. We first incubated 0.1 μM CAII with 1.0 μM SA–DNA for 30 min to allow for sufficient binding of SA–DNA to CAII. We then added 1.0 μM NHS-DNA and varying concentrations of free SA (0, 5, 25, 125 μM). The excess amounts of free SA could compete with SA/NHS-DNA hybrids and interfere with the process of association and dissociation of affinity binding. Interference of the competing free SA resulted in the incomplete conjugation (Fig. S7, ESI†). These results support that the iterative process of affinity interaction and covalent binding is the underlying reason of the complete conjugation. We have also conducted mass spectrometry analysis and confirmed the 1:1 stoichiometry of the protein:DNA conjugate (Fig. S8, ESI†).
We further examined whether other types of affinity ligands can be used to accomplish quantitative conjugation. We chose thrombin as the target for DNA conjugation because its two aptamers, with Kd of ∼100 nM and 0.5 nM,33–35 allow for testing the effect of binding affinity on the conjugation. We extended each aptamer at its 3′ end with the same DNA sequence used for the CAII study (Table S1, ESI†), to attach the same NHS moiety. When the ratio of DNA:thrombin was 1:1, the conjugation yields of 39% and 31% were achieved using TBA (Fig. S9, ESI†) and HD22 (Fig. S10, ESI†) as the affinity aptamers, respectively. When the ratio of DNA:thrombin increased to 10:1 (lane 2 and 4, Fig. 2c), complete conjugation was achieved with the use of both aptamers. Taken together with the results on the binding of SA to CAII (Kd of 3.2 μM), we have demonstrated quantitative conjugation using affinity ligands of diverse affinities, with Kd varying by ∼6400 folds.
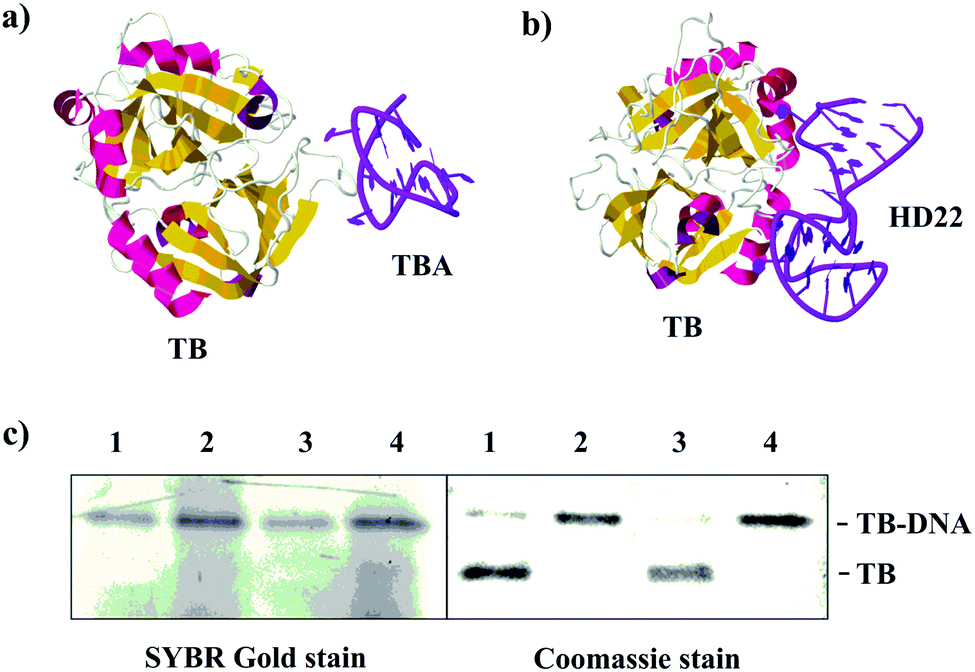 | ||
| Fig. 2 (a and b) Structures of human α-thrombin (TB) in complexes with its aptamers, TBA (PDB entry 4DII) and HD22 (PDB entry 4I7Y). (c) SDS-PAGE (12%) stained with Coomassie blue and SYBR Gold characterizing the conjugation products. Concentration of TB was kept at 0.1 μM. Lane 1, TBA/NHS-DNA at 0.2 μM; lane 2, TBA/NHS-DNA at 1 μM; lane 3, HD22/NHS-DNA at 0.2 μM; lane 4, HD22/NHS-DNA at 1 μM. | ||
We further used the CAII–DNA conjugate, without any purification, to build a molecular switch36,37 that either inhibits or restores the activity of CAII. To test the inhibition of CAII, we measured the fluorescence of 6-carboxyfluorescein, produced by the CAII-catalyzed hydrolysis of the fluorogenic substrate 6-carboxyfluorescein diacetate (CFDA) (Fig. 3a). The free SA was not effective to inhibit the activity of CAII (Fig. S12, ESI†). However, the activity of CAII was completely inhibited by using the SA–DNA and the CAII–DNA conjugate that we synthesized (Fig. 3b). This increase in enzyme inhibition was because the hybridization of the complementary sequences in SA–DNA and CAII–DNA facilitated the binding of SA to CAII (Fig. 3a, left schematic). To build a molecular switch, we introduced an initiator DNA that could remove the SA–DNA inhibitor from the CAII enzyme through a toehold-mediated strand-displacement reaction (Fig. 3a). Indeed, the addition of the initiator DNA increased the fluorescence (Fig. 3b) due to the restoration of the activity of the previously inhibited CAII enzyme. These results show that the activity of CAII–DNA could be effectively inhibited by SA–DNA and that this inhibition could be reversed, i.e., a molecular switch. Importantly, the construction of this molecular switch did not require any separation of the CAII–DNA conjugation product, demonstrating the benefit of the efficient conjugation of DNA to protein with a 1:1 stoichiometry.
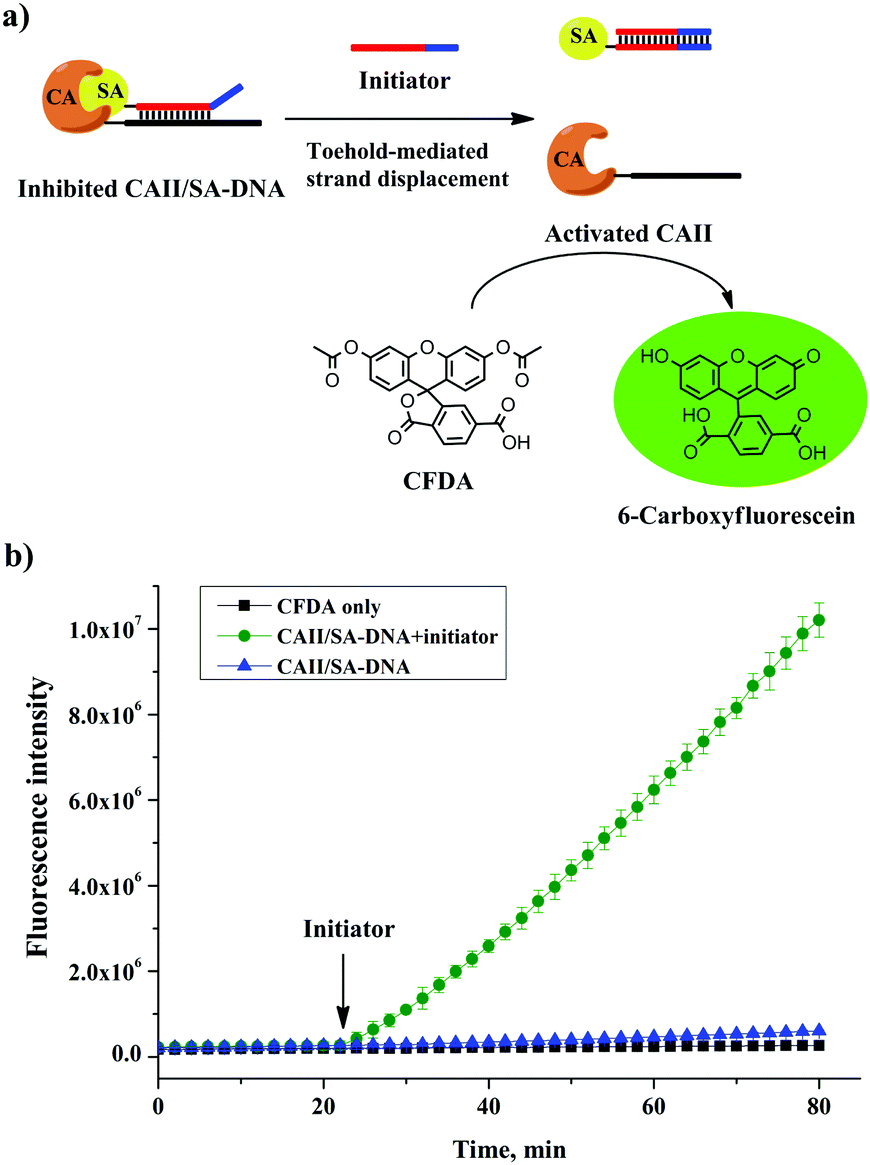 | ||
| Fig. 3 (a) Removal of the inhibitor SA from the CAII enzyme by an initiator DNA through a toehold-mediated strand displacement reaction. (b) Real-time monitoring of fluorescence produced from the CAII-catalyzed reaction shown in (a), in response to the initiator DNA. | ||
In summary, we have demonstrated the quantitative conjugation of DNA to specific proteins with a precise 1:1 stoichiometry. Efficient conjugation of DNA to carbonic anhydrase II and α-thrombin, using affinity ligands with dissociation constants varying by ∼6400 folds, indicates the generality of our strategy. Without the need for the separation of DNA–protein conjugates, we envision diverse applications, such as in biosensing, protein identification and quantification, targeted drug delivery, and manipulation of cell function.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Boozer, J. Ladd, S. Chen and S. Jiang, Anal. Chem., 2006, 78, 1515 CrossRef PubMed.
- J. Fu, M. Liu, Y. Liu, N. W. Woodbury and H. Yan, J. Am. Chem. Soc., 2012, 134, 5516 CrossRef PubMed.
- J. Fu, Y. R. Yang, A. Johnson-Buck, M. Liu, Y. Liu, N. G. Walter, N. W. Woodbury and H. Yan, Nat. Nanotechnol., 2014, 9, 531 CrossRef PubMed.
- Z. Zhao, J. Fu, S. Dhakal, A. Johnson-Buck, M. Liu, T. Zhang, N. W. Woodbury, Y. Liu, N. G. Walter and H. Yan, Nat. Commun., 2016, 7, 10619 CrossRef PubMed.
- T. Sano, C. L. Smith and C. R. Cantor, Science, 1992, 258, 120 CrossRef PubMed.
- S. Fredriksson, M. Gullberg, J. Jarvius, C. Olsson, K. Pietras, S. M. Gústafsdóttir, A. Ostman and U. Landegren, Nat. Biotechnol., 2002, 20, 473 CrossRef PubMed.
- Y. Xiang and Y. Lu, Nat. Chem., 2011, 3, 697 CrossRef PubMed.
- H. Zhang, X. F. Li and X. C. Le, Anal. Chem., 2012, 84, 877 CrossRef PubMed.
- H. Zhang, F. Li, B. Dever, C. Wang, X. F. Li and X. C. Le, Angew. Chem., Int. Ed., 2013, 52, 10698 CrossRef PubMed.
- O. Söderberg, M. Gullberg, M. Jarvius, K. Ridderstråle, K. J. Leuchowius, J. Jarvius, K. Wester, P. Hydbring, F. Bahram, L. G. Larsson and U. Landegren, Nat. Methods, 2006, 3, 995 CrossRef PubMed.
- R. Jungmann, M. S. Avendaño, J. B. Woehrstein, M. Dai, W. M. Shih and P. Yin, Nat. Methods, 2014, 11, 313 CrossRef PubMed.
- F. Chen, P. W. Tillberg and E. S. Boyden, Science, 2015, 347, 543 CrossRef PubMed.
- R. Jungmann, M. S. Avendaño, M. Dai, J. B. Woehrstein, S. S. Agasti, Z. Feiger, A. Rodal and P. Yin, Nat. Methods, 2016, 13, 439 CrossRef PubMed.
- S. M. Douglas, I. Bachelet and G. M. Church, Science, 2012, 335, 831 CrossRef PubMed.
- C. M. Niemeyer, T. Sano, C. L. Smith and C. R. Cantor, Nucleic Acids Res., 1994, 22, 5530 CrossRef PubMed.
- M. Lovrinovic, L. Fruk, H. Schröder and C. M. Niemeyer, Chem. Commun., 2007, 353 RSC.
- C. M. Niemeyer, Angew. Chem., Int. Ed., 2010, 49, 1200 CrossRef PubMed.
- S. A. Kazane, D. Sok, E. H. Cho, M. L. Uson, P. Kuhn, P. G. Schultz and V. V. Smider, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 3731 CrossRef PubMed.
- S. Barbuto, J. Idoyaga, M. Vila-Perelló, M. P. Longhi, G. Breton, R. M. Steinman and T. W. Muir, Nat. Chem. Biol., 2013, 9, 250 CrossRef PubMed.
- S. I. Liang, J. M. McFarland, D. Rabuka and Z. J. Gartner, J. Am. Chem. Soc., 2014, 136, 10850 CrossRef PubMed.
- B. P. Duckworth, Y. Chen, J. W. Wollack, Y. Sham, J. D. Mueller, T. A. Taton and M. D. Distefano, Angew. Chem., Int. Ed., 2007, 46, 8819 CrossRef PubMed.
- M. Erkelenz, C. H. Kuo and C. M. Niemeyer, J. Am. Chem. Soc., 2011, 133, 16111 CrossRef PubMed.
- X. Li and D. R. Liu, Angew. Chem., Int. Ed., 2004, 43, 4848 CrossRef PubMed.
- G. Li, Y. Liu, Y. Liu, L. Chen, S. Wu, Y. Liu and X. Li, Angew. Chem., Int. Ed., 2013, 52, 9544 CrossRef PubMed.
- D. M. Bauer, A. Rogge, L. Stolzer, C. Barner-Kowollik and L. Fruk, Chem. Commun., 2013, 49, 8626 RSC.
- B. J. H. M. Rosier, G. A. O. Cremers, W. Engelen, M. Merkx, L. Brunsveld and T. F. A. de Greef, Chem. Commun., 2017, 53, 7393 RSC.
- C. B. Rosen, A. L. Kodal, J. S. Nielsen, D. H. Schaffert, C. Scavenius, A. H. Okholm, N. V. Voigt, J. J. Enghild, J. Kjems, T. Tørring and K. V. Gothelf, Nat. Chem., 2014, 6, 804 CrossRef PubMed.
- J. B. Trads, T. Tørring and K. V. Gothelf, Acc. Chem. Res., 2017, 50, 1367 CrossRef PubMed.
- J. L. Vinkenborg, G. Mayer and M. Famulok, Angew. Chem., Int. Ed., 2012, 51, 9176 CrossRef PubMed.
- R. Wang, D. Lu, H. Bai, C. Jin, G. Yan, M. Ye, L. Qiu, R. Chang, C. Cui, H. Liang and W. Tan, Chem. Sci., 2016, 7, 2157 RSC.
- C. Cui, H. Zhang, R. Wang, S. Cansiz, X. Pan, S. Wan, W. Hou, L. Li, M. Chen, Q. Liu and W. Tan, Angew. Chem., Int. Ed., 2017, 56, 11954 CrossRef PubMed.
- V. Krishnamurthy, G. K. Kaufman, A. R. Urbach, I. Gitlin, K. L. Gudiksen, D. B. Weibel and G. M. Whitesides, Chem. Rev., 2008, 108, 946 CrossRef PubMed.
- L. C. Bock, L. C. Griffin, J. A. Latham, E. H. Vermaas and J. J. Toole, Nature, 1992, 355, 564 CrossRef PubMed.
- D. M. Tasset, M. F. Kubik and W. Steiner, J. Mol. Biol., 1997, 272, 688 CrossRef PubMed.
- B. Deng, Y. Lin, C. Wang, F. Li, Z. Wang, H. Zhang, X. F. Li and X. C. Le, Anal. Chim. Acta, 2014, 837, 1 CrossRef PubMed.
- A. Saghatelian, K. M. Guckian, D. A. Thayer and M. R. Ghadiri, J. Am. Chem. Soc., 2003, 125, 344 CrossRef PubMed.
- W. Engelen, B. M. Janssen and M. Merkx, Chem. Commun., 2016, 52, 3598 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, gel images and mass spectra. See DOI: 10.1039/c8cc03268h |
| This journal is © The Royal Society of Chemistry 2018 |