
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWeakly-coordinating N-oxide and carbonyl groups for metal-catalyzed C–H activation: the case of A-ring functionalization
Eufrânio N.
da Silva Júnior
 *a,
Guilherme A. M.
Jardim
a,
Roberto S.
Gomes
bc,
Yu-Feng
Liang
d and
Lutz
Ackermann
*d
*a,
Guilherme A. M.
Jardim
a,
Roberto S.
Gomes
bc,
Yu-Feng
Liang
d and
Lutz
Ackermann
*d
aInstitute of Exact Sciences, Department of Chemistry, Federal University of Minas Gerais, Belo Horizonte, 31270-901, MG, Brazil. E-mail: eufranio@ufmg.br; Web: http://www.eufraniolab.com Tel: +55 31 34095720
bFaculty of Exact Sciences and Technologies, Federal University of Grande Dourados, Dourados, MS, Brazil
cDepartment of Chemistry and Chemical Biology, Harvard University, Cambridge, Massachusetts 02138, USA
dInstitut für Organische und Biomolekulare Chemie, Georg-August-Universität Göttingen, Tammannstraße 2, Gottingen 37077, Germany. E-mail: lutz.ackermann@chemie.uni-goettingen.de; Tel: +49 551 393202
First published on 14th May 2018
Abstract
Compounds featuring weakly-coordinating N-oxides or carbonyl groups, as for instance, quinoline N-oxide and quinonoid systems represent important structural scaffolds with potential biological activities. Due to their biological importance, significant efforts have been devoted to devise robust methods for their step-economical preparation. Among these approaches, the C–H activation strategy has emerged as a powerful, versatile and efficient tool in molecular sciences. This feature article summarizes recent key advances in transition-metal-catalyzed C–H functionalization for A-ring functionalization of heterocyclic and quinoidal compounds by challenging weakly-coordinating entities, published prior to May 2018.
 Eufrânio N. da Silva Júnior | Eufrânio N. da Silva Júnior obtained his PhD in Chemistry from the University of Brasília in 2009. In 2010, he started his independent career as a Professor at the Federal University of Minas Gerais (UFMG). He is a member of the SBQ and RSC. He has received the RSC/BMOS Young Investigator Award (2015), Jones Travelling Fellowship (2016) and Capes-Humboldt Research Fellowship for experienced researchers (2018). His research interests are focused on C–H bond activation reactions, organocatalysis, mechanistic investigations, and synthesis of heterocyclic and naphthoquinoidal bioactive and fluorescent compounds. He has 76 publications related to Organic Chemistry and Medicinal Chemistry. |
 Guilherme A. M. Jardim | Guilherme A. M. Jardim received his degree in Chemistry in 2012. In 2014 he concluded his MSc at the Federal University of Minas Gerais under the supervision of Prof. Eufrânio N. da Silva Júnior and in the same year he started his PhD studies in the da Silva Júnior group working with new methods involving C–H bond activation for functionalization of quinoidal bioactive compounds. His research interests are focused on C–H bond functionalization and cross coupling reactions. He is also interested in Medicinal Chemistry aiming at the synthesis of bioactive compounds. |
 Roberto S. Gomes | Roberto S. Gomes obtained his PhD in Organic Chemistry from the University of São Paulo in 2011. Afterward, he joined the group of Prof. Adilson Beatriz at the Federal University of Mato Grosso do Sul as a postdoctoral assistant to design new chemical methodologies for the synthesis of biologically active compounds. In 2013, he started his independent career as an Assistant Professor at the Federal University of Grande Dourados. His research interests are focused on C–C bond activation and cross-coupling reactions. Currently, he is a Visiting Professor at Harvard University in association with Prof. E. J. Corey's Research Group. |
 Yu-Feng Liang | Yu-Feng Liang received his BS and MS degrees in 2009 and 2012 under the supervision of Prof. Hua-Jian Xu from Hefei University of Technology in China. Subsequently, he joined Prof. Ning Jiao's group at Peking University working with oxygenation via C–H/C–C bond activation, and obtained his PhD degree in 2015. Since 2016, he has been conducting his postdoctoral research at Georg-August-University Göttingen with Prof. Lutz Ackermann, as an Alexander von Humboldt Postdoctoral Fellow. His research interests are focused on transition metal catalyzed C–H and C–C bond functionalizations. |
 Lutz Ackermann | Lutz Ackermann studied Chemistry at the Christian-Albrechts-University Kiel, Germany, and received his PhD in 2001 for research under the supervision of Prof. Dr Alois Fürstner from Max-Plank-Institut für Kohlenforschung in Mülheim/Ruhr. He was a postdoctoral coworker in the laboratory of Prof. Dr Robert G. Bergman at the University of California at Berkeley before initiating his independent research programme in 2003 at the Ludwig Maximilians-University München supported by the Emmy Noether-programme of the DFG. In 2007, he was appointed as a Full (W3) Professor at the Georg-August University Göttingen. His recent awards and distinctions include an ERC Grant (2012) and a Gottfried-Wilhelm-Leibniz-Preis (2017) as well as visiting professorships at the Università degli Studi di Milano, the University of Wisconsin at Madison, the Università di Pavia, Osaka University, the Ecole supérieure de Physique et de Chimie Industrielles de la ville de Paris, the Università degli Studi di Perugia, and Kyoto University. The development and application of novel concepts for sustainable catalysis constitute his major current research interests, with a current topical focus on C–H activation. |
1. Introduction
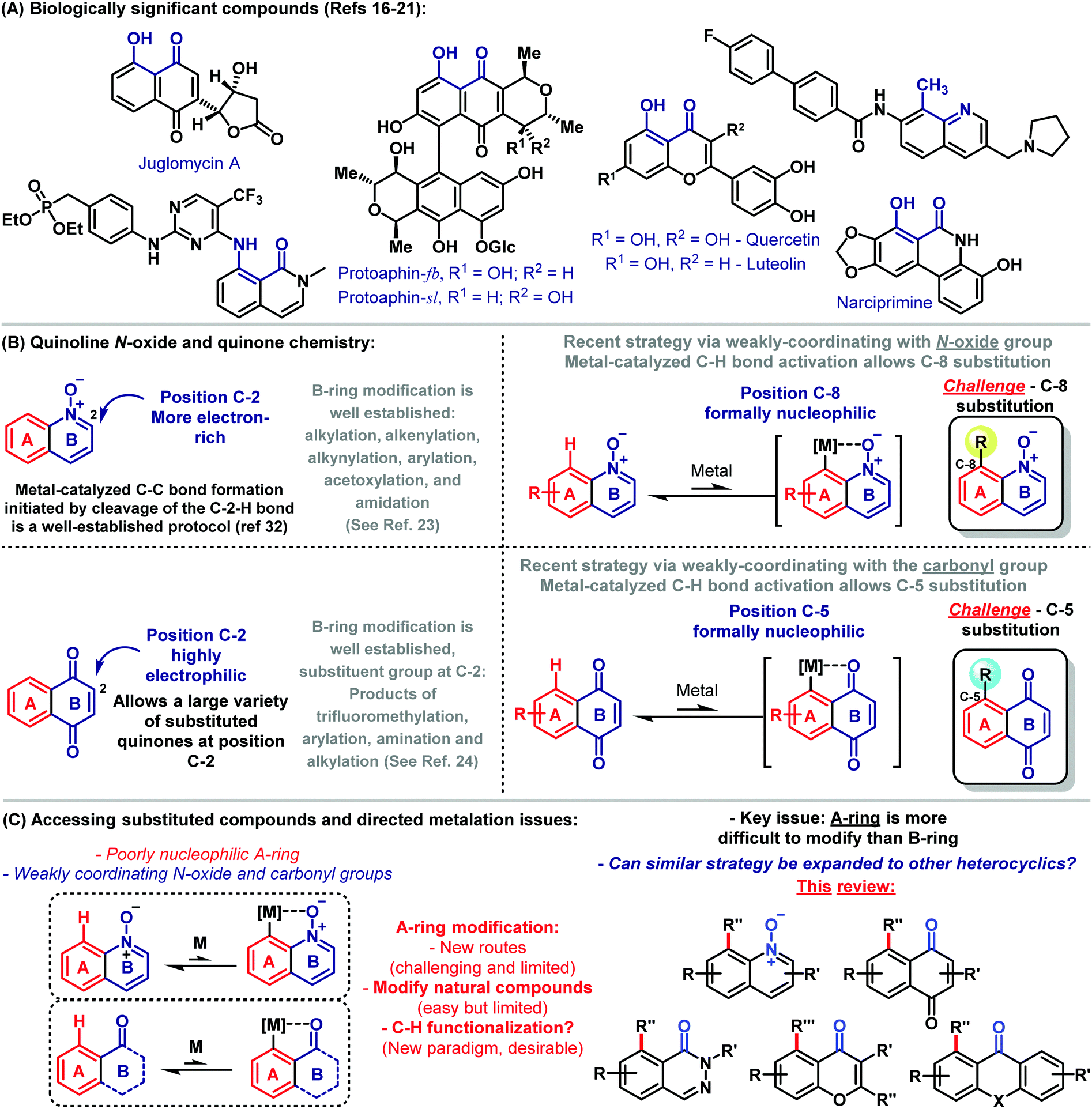
Since synthetic organic chemists have deciphered important aspects associated with the basis of molecular transformations, the synthetic community has developed efficient and versatile strategies aimed at the preparation of complex molecules with the fewest synthetic steps, in high yields and attending to several factors that make chemical synthesis truly innovative.1 In this sense, no approach in molecular synthesis has arguably become as revolutionary as C–H activation chemistry.2 Early examples of directed C–H functionalizations were described by inter alia Löffler,3 Corey,4 Barton,5 Bergman6 as well as Murai,7 and since then, this important field of science has been rapidly evolving. Nowadays, C–H functionalization represents an increasingly viable tool for the synthesis of complex molecules, occupying a space at the forefront of scientific exploration.8 For instance, a simplified route for the assembly of arylomycins was elegantly devised by means of C–H functionalization logic.9 Thus, a strategy was realized that mimics the putative biosynthesis through a copper-mediated oxidative phenol coupling.9 Methods aiming at directed C–H bond activation, functionalization of heterocycles, cascade reactions incorporating C–H functionalization and allylic C–H oxidation are among the challenges covered in this field and represent the potential of these reactions as a powerful tool in synthetic organic chemistry.10The control of the regioselectivity in C–H functionalization is critical for taking the full advantage of this important approach. By the correct choice of the catalyst, ligands, solvents and fine tuning of the reaction conditions, it is hence possible to control the position-diversity of the formed product.11 An important factor associated with the control of the reaction is also the use of a directing group (DG). In general, the coordination of a DG to a metal catalyst is the key for the site-selective functionalization of a specific C–H bond by proximity-induced chelation assistance. As recently discussed by Dong, endo-DGs are capable of forming an endocyclic π-bond after C–H metalation. C–H activations assisted by this sort of DGs have been widely studied.12 The diverse viable functional groups used as DGs include amides, amines, anilines, heterocyclic compounds, carboxylic acids, N-oxide and carbonyl groups.13 Among these DGs, the use of weakly-coordinating directing groups14 emerged as particularly important in order to expand the scope of the previously unavailable substrates. The control of the chemistry related to weakly-coordinating directing groups allows development of synthetic methods being useful for the preparation of heterocyclics and quinoidal compounds, which however bear challenges as to the coordination to the metal catalyst.15
Bioactive naturally occurring compounds, such as juglomycin A,16 protoaphin-fb and protoaphin-sl,17 quercetin,18 luteolin,19 and narciprimine,20 among others21 have hydroxyl, methyl or amine groups adjacent to a weakly-coordinating carbonyl group or potentially N-oxide group for metal-catalyzed C–H activation (Scheme 1A). Despite major advances related to C–H functionalizations, site- and chemoselective methods for direct catalytic modifications of complex heterocyclic scaffolds with weakly-coordinating DGs remain a key challenge.
 | ||
| Scheme 1 Overview. | ||
Quinoline N-oxide and quinoidal compounds are important scaffolds with potential biological activities.22 Because of their biological importance, significant efforts have been devoted to developing modern synthetic methods for their efficient assembly and diversification.23,24 In general, B-ring modification is reasonably well established, while protocols that allow for the direct functionalization of the A-ring are less explored (Scheme 1B).
Recent strategies via weakly-coordinating directing groups were used for metal-catalyzed C–H bond reactions and enabled the preparation of C-8 and C-5 substituted quinoline N-oxides and naphthoquinoidal compounds, respectively. This strategy arises as a gateway to functionalized derivatives. Herein, we discuss key progress for the synthesis of A-ring substituted quinoidal compounds and heterocyclics through transition metal-catalyzed C–H functionalization reactions (Scheme 1C).
2. Quinoline A-ring functionalization: N-oxide as a weakly-coordinating directing group
The development of new synthetic procedures for the facile modification of quinolines are of utmost importance in organic chemistry, since this motif can be found in a large number of natural and synthetic compounds.25 Most of the known C–H activation processes in quinolines allow for the functionalization at the C-2 position,23,26 but the simple conversion of quinolines into quinoline N-oxides expands the reaction arsenal for this core modification, making regioselectivity in positions other than C-2 possible.27 Among these, control of the selectivity on the carboxylic ring (C-8 position) is highly desirable, once the corresponding quinolines present important utilities in several areas.28The importance of the N-oxide group in regioselectivity lies in the fact that the nucleophilicity at the C-2 position is high enough for the reaction to occur via the electrophilic aromatic substitution (SeAr) pathway; in this position, there is poor participation of the directing group (DG). On the other hand, the metal center has to be directed into the site with low reactivity (C-8 position) and the most probable mechanistic route to this transformation is base assisted C–H metalation.29
Insights into C-2 and C-8 selective arylation of quinoline N-oxides were reported by Larionov and collaborators in an attempt to explain the site selectivity when palladium catalyzed C–H activation is employed.30 The reaction was carried out in thermal mode and under microwave irradiation, presenting broad scope and excellent yields with high C-8 regioselectivity. Analysis of several mechanistic scenarios via DFT calculations showed that acetic acid is essential for C-8 site selectivity by stabilizing the metalated theorized species (Scheme 2).
 | ||
| Scheme 2 C-8 selective arylation of quinoline N-oxides reported by Larionov.30 | ||
Following the same approach, Larionov and co-workers also described insights into the experiment and mechanism of palladium-catalyzed oxidative C8-selective C–H homocoupling of quinoline N-oxides.31 Using the same conditions for the C-8 arylation with major tuning of the reactant loading and exchanging Ag3PO4 with AgOAc, it was possible to achieve a homocoupling reaction, affording substituted biquinolyl N,N′-dioxides in good to excellent yields (Scheme 3).
 | ||
| Scheme 3 Biquinolyl N,N′-dioxides described by Larionov.31 | ||
In a remarkable report in 2014, Shibata and Matsuo described a direct alkenylation in the C-8 position catalyzed by a cationic rhodium(I) catalyst/xylylBINAP system.32 It was shown that the reaction proceeded in good to excellent yields and with high regioselectivity E/Z, with a broad scope of diarylacetylenes and quinoline N-oxides. Mechanistic studies involving deuterium labelling experiments with D2O showed that both C-2 and C-8 positions were surprisingly deuterated in the same ratio, pointing that equilibria between metalated species at C-2 and C-8 exist, with the predominance of the last one (Scheme 4).
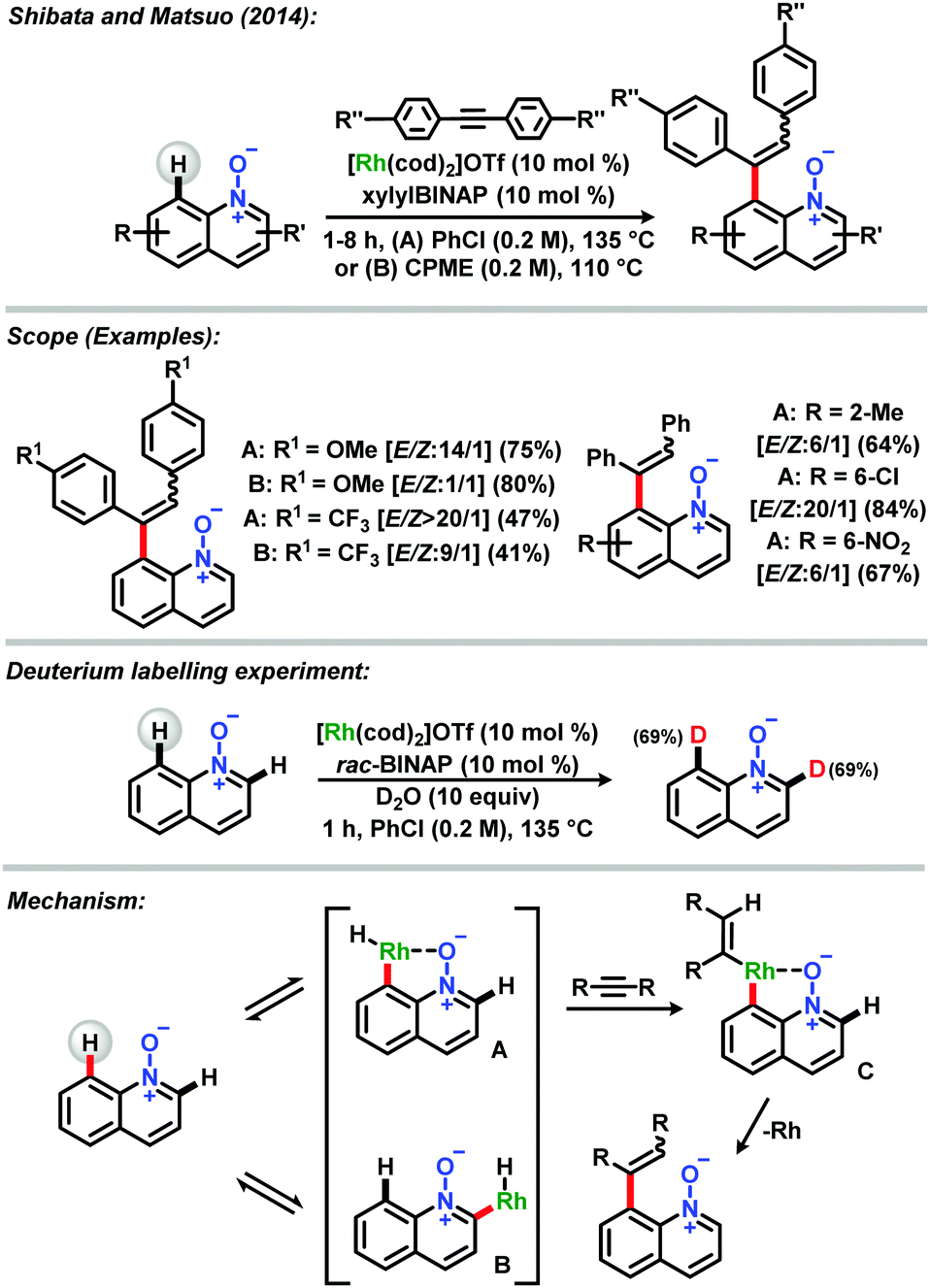 | ||
| Scheme 4 C-8 alkenylation catalyzed by a cationic rhodium(I) catalyst/xylylBINAP system described by Shibata and Matsuo.32 | ||
In the same year, Chang and co-workers reported both C-8 iodination and amidation of quinoline N-oxides, utilizing similar catalytic systems (Scheme 5).33 For C–H iodination, rhodium(III)/AgNTf2 was used together with NIS in 1,2-DCE under mild conditions to afford products in good to excellent yields. For the amidation reactions, rhodium(III) was exchanged with iridium(III) and tosyl azides were used as the amide source, with AcOH as an additive. The reactions were also carried out under the same mild conditions, leading to amide products in good to excellent yields. Mechanistic studies involving the synthesis and X-ray crystallographic analysis for the characterization of an isolable intermediate (A) showed the first example of a discrete iridacycle bound to N-oxide (Scheme 6).
 | ||
| Scheme 5 C-8 iodination and amidation of quinoline N-oxides.33 | ||
 | ||
| Scheme 6 Mechanistic studies for C-8 iodination and amidation of quinoline N-oxides reported by Chang.33 | ||
In another notable work, Chang reported the development and application of Ir(III)Cp* catalyzed C–H arylation using aryldiazonium tetrafluoroborates in an external oxidant-free approach.34 The current C–H arylation protocol was successfully employed in a wide range of substituted quinoline N-oxides and a broad scope of aryldiazonium tetrafluoroborates in moderate to good yields (Scheme 7).
 | ||
| Scheme 7 C–H arylation of quinoline N-oxides with aryldiazonium tetrafluoroborates reported by Chang.34 | ||
Sundararaju and co-workers described the C-8 selective allylation of quinoline N-oxides by achieving β-hydroxy and β-hydride elimination products selectively, using two different metals from group 9 under the same reaction conditions.35 Using a cobalt(III) catalyst, the allylation proceeded via β-hydroxy elimination at the C-8 position in good yields and with regioselectivity. Interestingly, exchanging the metal catalyst with a rhodium(III) complex caused the reaction to follow a different path, leading to the formation of β-aryl ketones via β-hydride elimination. Insights based on DFT studies showed that the energy required for β-hydroxy elimination is comparatively lower with cobalt(III) than with rhodium(III), this being the principal motive that drives the reaction with rhodium(III) to occur via β-hydride elimination (Scheme 8).
 | ||
| Scheme 8 C-8 selective allylation of quinoline N-oxides described by Sundararaju.35 | ||
The Sundararaju group also reported an efficient, scalable, atom-economical, regioselective air stable Co(III)Cp* C–H activation reaction between quinoline N-oxides and internal alkynes.36 The methodology tolerates various functional groups and dispenses the use of additives, and several symmetrical and unsymmetrical alkynes were employed with high selectivity. Curiously, oxygen atom transfer (OAT) was observed between the N–O directing group and the alkyne (Scheme 9).
 | ||
| Scheme 9 Reaction of quinoline N-oxides and internal alkynes catalysed by Co(III)Cp* described by Sundararaju.36 | ||
Liu and co-workers reported Cp*Rh(III)-catalyzed directed C–H methylation and arylation of quinoline N-oxides at the C-8 position using commercially available organotrifluoroborates as reagents.37 The methodology presents good regioselectivity, mild conditions and broad functional group tolerance, with a high scope of N-oxides. The methylated and arylated quinoline N-oxides were obtained in good to excellent yields, even in gram-scale reactions. Mechanistic studies aimed for the synthesis and characterization of the isolable rhodacycle intermediate A by the reaction of the N-oxide with stoichiometric amounts of [RhCp*Cl2]2, followed by the reaction with the substrates to afford the methylated N-oxide in 84% yield, showing that the five-membered rhodacycle could be a key intermediate formed via coordination of the rhodium catalyst with the O atom from the N-oxide moiety. This intermediate could be undergoing electrophilic C–H bond cleavage at the C-8 position, followed by transmetallation with the organotrifluoroborate and reductive-elimination to afford the corresponding products (Scheme 10).
 | ||
| Scheme 10 C–H methylation and arylation of quinoline N-oxides at the C-8 position reported by Liu.37 | ||
Despite the rapidly growing development of reactions involving C–H activation with quinoline N-oxide, the synthesis of this sort of molecules substituted at the C-8 position is quite new and advances in this area of research still need to be explored more deeply. In particular, catalytic systems that allow for the control of the regioselectivity aiming to select the regiodiversity of the formed product are still required. In this study, we aimed to draw attention to the already developed catalytic systems for quinoline A-ring functionalization by using N-oxide as a weakly-coordinating directing group. The challenges of discovering the adequate reaction conditions for the synthesis of quinolines substituted at position C-8 are reflected in the paucity of reported examples in the literature. The use of the N-oxide group as a DG for direct functionalization at C-8 will reflect on the synthesis of molecules with a complex structural framework via a modern and practical approach, from the C–H bond activation point of view.
3. Benzenoid A-ring modification of quinonoid systems: carbonyl as a weakly-coordinating directing group
First efforts aiming at the direct functionalization of the benzenoid ring of quinonoid systems utilizing carbonyl as a directing group can be traced back to 2009 in the work of Kakiuchi and co-workers, which described the direct C–H arylation of anthraquinone with arylboronates utilizing RuH2(CO)(PPh3)3 as a catalyst.38 Kakiuchi also proved later in 2012 that the same catalyst could provide the functionalization of anthraquinones with olefins via regioselective C–H alkylation. The catalyst also promoted C–O arylation with organoboronates, resulting in a pioneer chemoselective tandem C–H alkylation/C–O arylation reaction (Scheme 11).39 | ||
| Scheme 11 Direct functionalization of the benzenoid ring with carbonyl as a DG described by Kakiuchi.38,39 | ||
Despite the broad occurrence of napthoquinones in nature, direct benzenoid A-ring modifications of these motifs are often rare. In a pioneering report, Zhang and co-workers described a naphthoquinone-directed C–H annulation/C(sp3)–H bond cleavage reaction for the synthesis of tetracyclic naphthoxazoles.40 The methodology consisted in the use of [RhCp*Cl2]2/AgSbF6 as the catalytic system allied with stoichiometric equivalents of Cu(OAc)2·H2O. Such a reaction made the synthesis of a broad range of tetracyclic naphthoxazoles possible in good to excellent yields. The authors had described the presence of an electron donating group at position 2 of the naphthoquinoidal skeleton as an important strategy for enhancing the coordinating capacity of the directing group. The mechanism of the reaction involves the tautomerization of the quinone which then undergoes rhodium(III)-catalyzed C–H activation to form a five-membered rhodacycle (A) following by insertion of alkyne and subsequent reductive elimination. After cyclization, followed by oxidative aromatization the desired product was obtained. Zhang's procedure could be considered the milestone in C–H functionalization of naphthoquinones as the first case of benzenoid ring functionalization in these systems (Scheme 12).
 | ||
| Scheme 12 Synthesis of tetracyclic naphthoxazoles via C–H annulation/Csp3–H bond cleavage reaction by Zhang.40 | ||
Zhang continued to explore the reactivity of 2-amino-1,4-naphthoquinones towards the [RhCp*Cl2]2/AgSbF6 system, applying as electrophilic acrylates.41 This time, only small amounts of oxidant (Cu(OAc)2·H2O, 20 mol%) were sufficient to promote C-5 functionalization. The reaction proceeded smoothly with other olefins with sulfur and phosphorous based substituents, as well as a broad range of amino substituents at the C-2 position. Mechanistic insights allied with deuterium labelled experiments showed that a possible intermediate would be a five membered rhodacycle and C5–H bond cleavage was involved in the rate determining step (Scheme 13).
 | ||
| Scheme 13 Substituent-enabled C-5 oxidative dehydrogenative cross-coupling of 1,4-naphthoquinones with alkenes described by Zhang.41 | ||
In 2016, the da Silva Júnior and Bower groups reported a rhodium(III) catalytic system for direct C–H activation at the C-5 position of the naphthoquinoidal system providing an access to A-ring substituted analogues.42 The methodology made the synthesis of C-5 halogenated naphthoquinones possible in good yields and high regioselectivity and functional group tolerance, being considered an important method for direct naphthoquinoidal functionalization. The reaction consisted of the combination of [RhCp*Cl2]2/AgNTf2 as the catalytic system, stoichiometric amounts of Cu(OAc)2 as the oxidant and NIS as the source of iodine, allied with microwave conditions to increase yields and lower the reaction time. The authors believed in the hypothesis that the rhodacycle intermediate A could exist in low concentrations in equilibrium with the naphthoquinoidal moiety, being formed via a base-assisted metalation pathway directed by the weak coordination of the carbonyl group, and “fast trapping” of this intermediate with a highly reactive source of iodine like NIS could provide the desired products (Scheme 14).
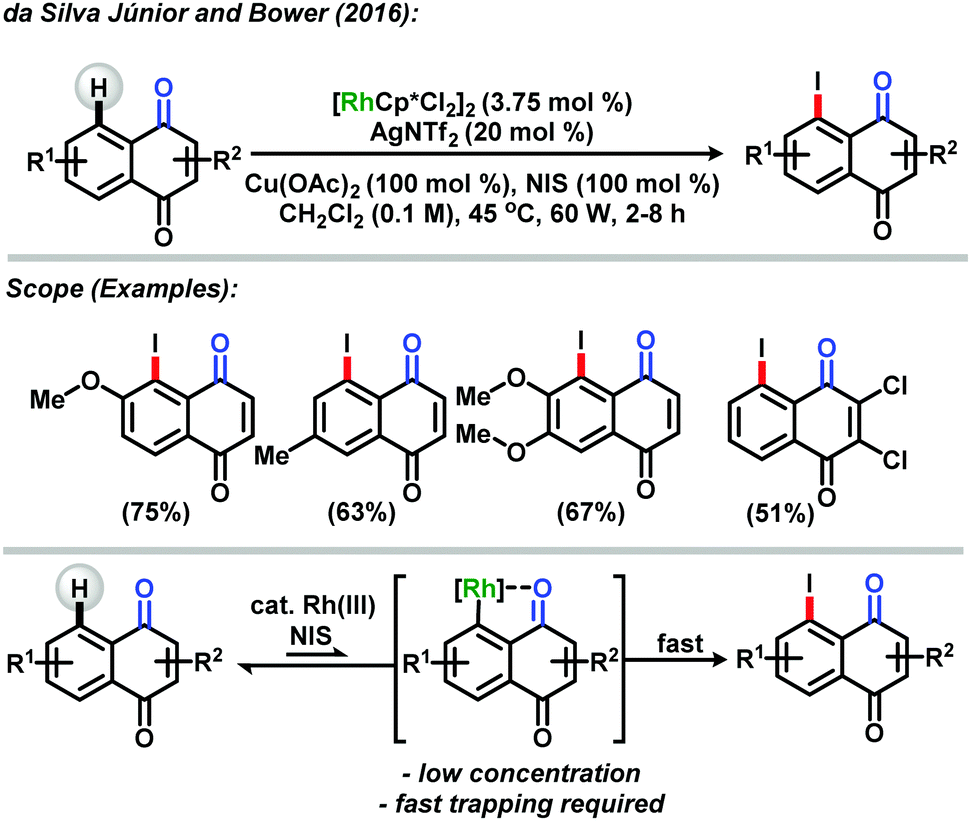 | ||
| Scheme 14 Rhodium(III)-catalyzed A-ring direct C–H iodination of naphthoquinones by da Silva Júnior and Bower.42 | ||
The insertion of a versatile atom like iodine opened way for the modification of the naphthoquinoidal moiety, as shown by da Silva Júnior and Bower in different works.42,43 Use of palladium cross-coupling reactions proved to be an efficient tool to access novel C-5 functionalized naphthoquinoidal scaffolds with remarkable trypanocidal activity (Scheme 15).
 | ||
| Scheme 15 Further modifications of the A-ring of naphthoquinones reported by da Silva Júnior and Bower.42,43 | ||
In 2018, the da Silva Júnior group also reported the direct sequential C–H iodination/organoyl-thiolation for benzenoid A-ring modification of naphthoquinones.44 The use of rhodium(III) catalyzed C–H iodination resulted in the development of a new protocol based on a copper(I) catalyzed reaction between the iodinated naphthoquinones and AgSR salts to provide the corresponding thiolated naphthoquinones in excellent yields. The new methodology proved to be highly efficient with most yields up to 90%. The derivatives were tested against Trypanosoma cruzi strains and presented high in vitro activity against the parasite (Scheme 16).
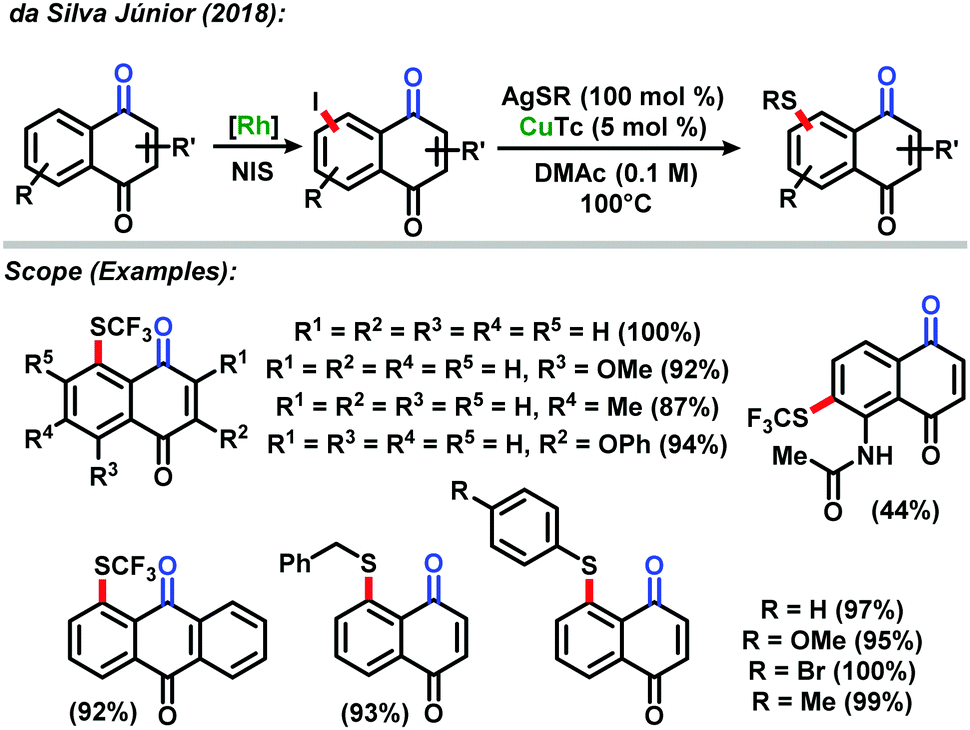 | ||
| Scheme 16 Direct sequential C–H iodination/organoyl-thiolation of naphthoquinones described by da Silva Júnior.44 | ||
In 2015, You and co-workers reported standardization of a rhodium(III)-catalysed cyclization method to obtain 1,8-dioxapyrenes and 1,12-dioxaperylenes from 1,4-naphthoquinones and 9,10-phenanthraquinones, respectively (Scheme 17).45 In this investigation 1,4-naphthoquinone and 1,2-diphenylacetylene were used as model substrates for evaluating the cyclisation process with [RhCp*Cl2]2 as the catalyst, AgSbF6 and Cu(OAc)2 as the oxidizing agent.
 | ||
| Scheme 17 1,8-Dioxapyrenes and 1,12-dioxaperylenes from 1,4-naphthoquinones and 9,10-phenanthraquinones described by You.45 | ||
With the optimized reaction conditions in hand, the authors extended the scope of 1,4-naphthoquinone and alkyne substrates. A wide range of 2-substituted 1,4-naphthoquinones smoothly underwent cyclization with 1,2-diphenylacetylene to afford 1,8-dioxapyrenes in moderate to good yields. Several halogenated 1,2-diarylacetylenes were also tested and the desired compounds were obtained in good yields. An asymmetrical alkyne was also evaluated under the same reaction conditions using 1,4-naphthoquinone as the substrate, when dioxane was employed as the solvent and PivOH as an additive, furnishing a regioselective product. In the case of the cyclisation reaction with terminal alkynes and dialkyl alkynes the reaction condition was not suitable to afford the desired compounds.
The cyclisation of 9,10-phenanthraquinones with alkynes was also explored. Under the optimal condition, several 9,10-phenanthraquinones and 1,2-diarylacetylenes underwent conversion to their corresponding 1,12-dioxaperylene derivatives in moderate to good yields (Scheme 17).
The use of naphthoquinoidal compounds for C–H bond activation with carbonyl as a DG allowed the development of important synthetic methodologies for A-ring functionalization of bioactive systems. In general, the chemical reactivity of quinones is peculiar and demands more accurate studies for discovering appropriate conditions for the respective catalytic reactions described in this study. As discussed by the da Silva Júnior and Bower groups, the modification of naphthoquinones is limited by (a) their susceptibility to reduction, (b) their high electrophilicity, (c) the low nucleophilicity of the benzenoid A-ring and (d) the weak coordinating ability of the B-ring carbonyls.42 These factors make the search for new reactions involving quinoidal compounds more complex and help us to understand the shortage of examples of methods employing such C–H functionalizations as a strategy for the synthesis of benzenoid A-ring modified molecules. This work makes clear the need to develop new synthetic strategies with the use of catalytic systems accessible for the modification of the A-ring of quinones. Finding viable conditions for the use of cheaper and available transition metals, such as cobalt and others, is still a challenge in this rapidly developing field of research.
4. A-ring functionalization of heterocycles: quinolones, phthalazinones, chromones and others
Based on the methodologies presented in Sections 1 and 2, recently, diverse research groups have explored synthetic alternatives for the selective functionalization of heterocyclics such as quinolones, phthalazinones and chromones.Hong and co-workers described an efficient method for the C-5 alkynylation of 4-quinolones using a weakly coordinating carbonyl group as a DG, via rhodium catalyzed C–H activation.46 The use of the catalyst [RhCp*(MeCN)3(SbF6)2] promoted the coupling reactions without any additives, providing alkylynated quinolones in good to excellent yields using a highly versatile reagent TIPS-EBX (Scheme 18). The authors also demonstrated the potential application of the methodology developed for the synthesis of the oxygenated aaptaminoid derivative (A). Initially, a rhodium-catalyzed C5-selective alkynylation method was used to prepare the 4-quinolone used in the preparation of the compound (A) that was obtained via 5-exo-dig cyclization in moderate 73% yield.
 | ||
| Scheme 18 Method for C-5 alkynylation of 4-quinolones developed by Hong.46 | ||
Use of rhodium catalysis proved to be also efficient in the C-8 functionalization of isoquinolones as described by Patil and co-workers (Scheme 19).47 The authors provided an efficient and robust protocol for the synthesis of C-8-alkynylated isoquinolones, presenting a scope with good to excellent yields and a high range of synthetic useful functional groups like –F, –Cl, –Br, –CF3, among others. Again, TIPS-EBX proved to be useful for transformations involving weakly coordinating groups as directing ones.
 | ||
| Scheme 19 C-8 functionalization of isoquinolones as described by Patil.47 | ||
Authors have proposed the mechanism for C-8-selective C–H alkynylation of isoquinolones. They suggested that the first event would be the coordination of the catalyst to the heterocycle by C–H bond activation followed by a concerted metalation/deprotonation pathway leading to the intermediate with the metal coordinated to the carbonyl group. After this process the acetylene may coordinate to the rhodium and insertion of acetylene in the Rh–C bond can occur affording the intermediate which would finally lead to the formation of the C-8 substituted product and regeneration of the rhodium catalyst.
Patel and collaborators have developed a method for regiospecific C–H/O–H annulations of quinolones with internal alkynes catalyzed by a ruthenium complex.48 Initially, the authors have established the optimized reaction conditions for the annulation with subsequent exploration of the scope of the reaction. The coupling between several substituted heterocycles and symmetrical internal alkynes was studied. In general, the desired annulated products were prepared in moderate yields (Scheme 20).
 | ||
| Scheme 20 Regiospecific annulations of quinolones with internal alkynes described by Patel.48 | ||
In 2016, Huestis reported the use of rhodium(III)-catalyzed functionalization of 1-(2H)-phthalazinones at C-8.49 The author reported that the reactions of [Cp*Rh(MeCN)3](SbF6)2 in the presence of Cu(OAc)2 as an oxidant led to good yields of the exclusively desired C-8 alkenylated phthalazinone products (Scheme 21). In general, the acrylates can react including brominated phthalazinones to afford the desired product in 85% yield as well as sterically hindered compounds with the respective product in 47% yield. In the case of vinyl sulfones and vinyl sulfonates, excellent yields can be observed for alkenylated phthalazinone products. On the other hand, vinyl phosphonates and vinyl sulfonamide afforded the products in low yields. Halogenated substrates were also tested by using aromatic alkenes, such as styrene and 2-vinylnaphthalene. This method represents an unprecedented method for C–H functionalization of phthalazinones at C-8.
 | ||
| Scheme 21 C-8 functionalization of phthalazinones reported by Huestis.49 | ||
In the same manuscript,49 Huestis reported hydroarylation, C–H alkenylation and C–H iodination of 1-(2H)-phthalazinones at C-8 (Scheme 22). When phthalazinone is allowed to react with diphenylacetylene with a rhodium catalyst in the presence of acetic acid, trisubstituted alkenes can be obtained in moderate to good yields. The reaction also occurs in hindered phthalazinones and N-substituted compounds. The reaction using 1-phenyl-1-propyne generated a mixture of E/Z isomers (46% and 27% yields). Iodination of phthalazinones with optimized conditions provided the respective iodinated products in moderate yields. Additionally, 6-bromo- and 6-chloro-phthalazinones were iodinated to furnish the respective products in 55% and 32% yields, respectively. This method represents an important contribution for phthalazinone chemistry allowing the synthesis of C-8 substituted molecules by using a practical and selective method.
 | ||
| Scheme 22 C–H alkenylation and C–H iodination of 1-(2H)-phthalazinones at C-8 described by Huestis.49 | ||
Besides naphthoquinones, chromones and xanthones have attracted particular attention due to their diversified biological activities. In 2016, Kim and co-workers50 reported optimization of a reaction between several chromones, xanthones and N-methylmaleimide under rhodium-catalyzed conditions. The C–H activated coupling at the C-5 position using cationic rhodium(III) catalysis with pivalic acid (PivOH) additive afforded the desired targets in good to excellent yields (Scheme 23). The scope of this reaction showed that the rhodium-based coupling using chromones bearing electron donor groups at positions C-2 and C-3 afforded the desired compounds in good to excellent yields; however, chromone containing an electron-deficient substituent at the C8-position underwent the coupling reaction in a relatively lower yield which suggested that the cross-coupling reaction could follow an electrophilic rhodation pathway in the C–H cleavage step. Fluoro-substituted chromone at the C6-position was found to be a suitable substrate furnishing products in high yield (90%). Alkyl- and aryl-substituted maleimides smoothly participated in the C5-alkylation reaction to afford the products in high yields. In general, when xanthones were used, the products were obtained in moderate to excellent yields (Scheme 23).
 | ||
| Scheme 23 Heterocycles and N-methylmaleimide reactions under rhodium-catalyzed conditions described by Kim.50 | ||
Hong and co-workers have described ruthenium(II)-catalyzed hydroxylation of flavone and chromone derivatives.51 Naturally occurring flavonoid compounds, as for instance, luteolin19 represent a major class of biologically active substances. Inspired by the previous methodology described by the Rao and Ackermann groups52 for catalytic direct oxygenation of aromatic esters, amides and ketones, the Hong group demonstrated C-5 hydroxylation of flavones via ruthenium-based catalysis in the presence of PIFA as an oxidizing agent. The reaction well tolerates a wide scope of substituents. The mechanism of the reaction goes through the interaction of the metal with the carbonyl group that acts as a directing group. This reaction is another successful example of the use of the carbonyl group as a DG (Scheme 24).
 | ||
| Scheme 24 Hydroxylation of flavone and chromone derivatives reported by Hong.51 | ||
Antonchick and co-workers53 have reported direct oxidative cross-coupling at the C-5 position of chromones and flavones via catalysis with rhodium. The compounds were obtained in good to excellent yields using a broad range of alkenes for the respective reactions. The products were obtained with high regioselectivity. This reaction is an interesting example that describes the ability of chromones and flavones to coordinate with the transition metal better than the quinonoid system of the quinone. In this reaction type the quinone acts as an electrophile in the reaction. The mechanism of the reaction proposed by the authors is similar to that already discussed in other examples shown above (Scheme 25).
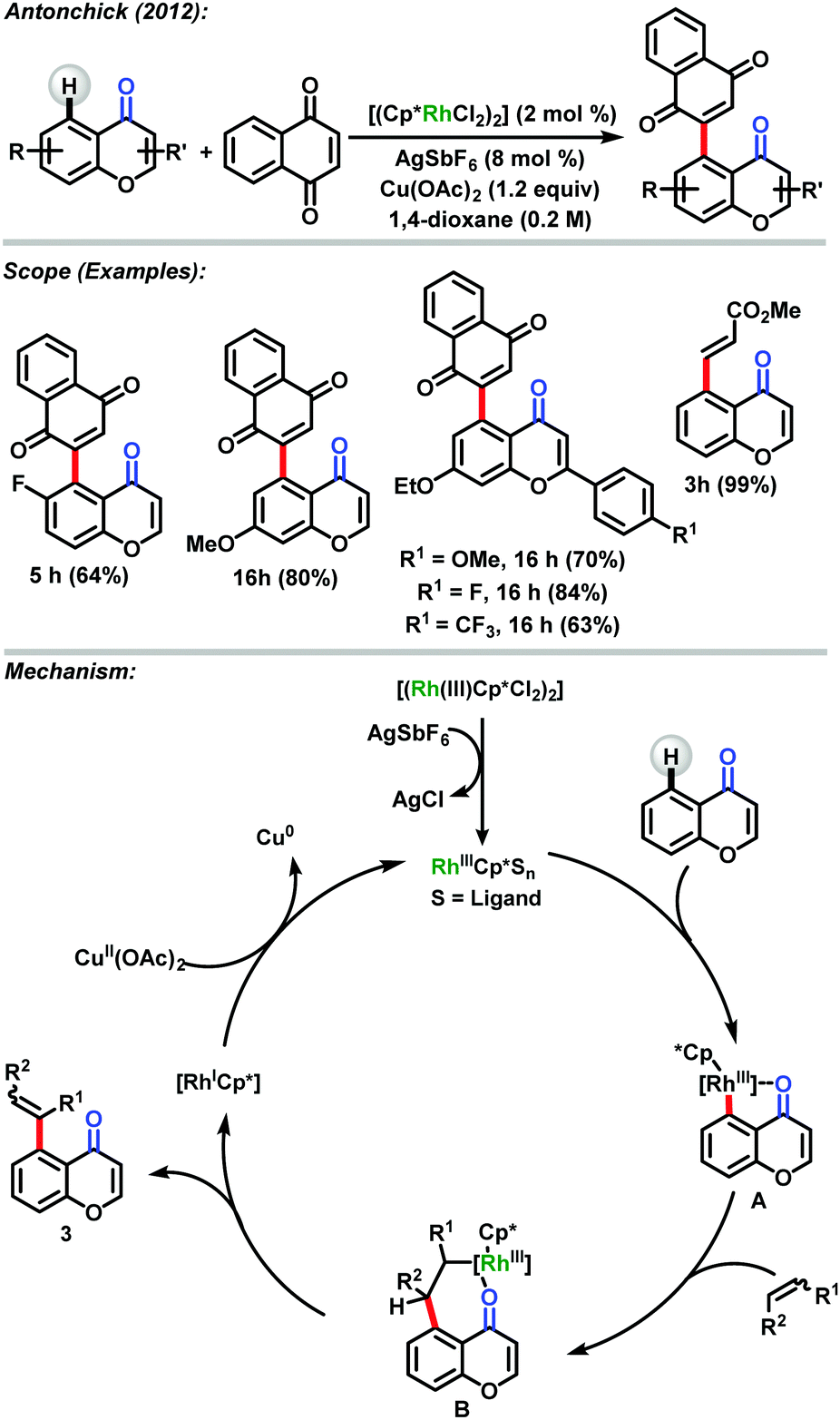 | ||
| Scheme 25 Direct oxidative cross-coupling at the C-5 position of chromones and flavones via catalysis with rhodium developed by Antonchick.53 | ||
This study reflects the importance of the carbonyl group as a directing group in C–H activation reactions. Different heterocyclics were functionalized via reactions involving transition metals. The examples described herein provide a basis for further research to be undertaken in order to develop synthetic methods for functionalizing other heterocyclics using a similar strategy.
5. Conclusions
The diversity of viable methodologies involving C–H activation is impressive. Despite major recent advances, weakly-coordinating N-oxide and carbonyl groups continue to be underdeveloped for metal-catalyzed C–H activation. The C–H functionalization arsenal used for the preparation of A-ring functionalized systems discussed in this review is based on transition metals, such as rhodium, palladium and ruthenium, with only rare examples using iridium or earth-abundant cobalt catalysts. Thus, this research arena is still underdeveloped, and we hope that our summary stimulates research in A-ring modifications of heterocyclic and quinoidal structural motifs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. N. da Silva Júnior acknowledges funding from CNPq (PVE 401193/2014-4, 404466/2016-8 and PQ 305741/2017-9), FAPEMIG (Edital 01/2014 APQ-02478-14 and Programa Pesquisador Mineiro – PPM X), CAPES, INCT-Catalysis and Capes-Humboldt research fellowship programme for experienced researchers (Proc. No. 88881.145517/2017-01). L. Ackermann acknowledges funding from the DFG (Gottfried-Wilhelm-Leibniz prize) and the Alexander von Humboldt Foundation (fellowship to Y. L.).References
- (a) K. C. Nicolaou and E. J. Sorensen, Classics in Total Synthesis: Targets, Strategies, Methods, Wiley, 1996 Search PubMed; (b) E. J. Corey and X.-M. Cheng, The Logic of Chemical Synthesis, John Wiley & Sons, Inc., 1995 Search PubMed.
- For recent reviews on transition metal-catalyzed C–H bond activation, see: (a) P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199 CrossRef; (b) W. Ma, P. Gandeepan, J. Li and L. Ackermann, Org. Chem. Front., 2017, 4, 1435 RSC; (c) G. Cera and L. Ackermann, Top. Curr. Chem., 2016, 374, 191 Search PubMed; (d) C. Borie, L. Ackermann and M. Nechab, Chem. Soc. Rev., 2016, 45, 1368 RSC; (e) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef PubMed; (f) T. Brueckl, R. D. Baxter, Y. Ishihara and P. S. Baran, Acc. Chem. Res., 2012, 45, 826 CrossRef PubMed; (g) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC; (h) Q. Wang, Y. Su, L. Li and H. Huang, Chem. Soc. Rev., 2016, 45, 1257 RSC; (i) B. Song and B. Xu, Chem. Soc. Rev., 2017, 46, 1103 RSC; (j) D.-S. Kim, W.-J. Park and C.-H. Jun, Chem. Rev., 2017, 117, 8977 CrossRef PubMed; (k) H. M. L. Davies and D. Morton, ACS Cent. Sci., 2017, 3, 936 CrossRef PubMed.
- K. Loffler and S. Kober, Ber. Dtsch. Chem. Ges., 1909, 42, 3431 CrossRef.
- E. J. Corey and W. R. Hertler, J. Am. Chem. Soc., 1958, 80, 2903 CrossRef.
- D. H. R. Barton, J. M. Beaton, L. E. Geller and M. M. Pechet, J. Am. Chem. Soc., 1960, 82, 2640 CrossRef.
- A. H. Janowicz and R. G. J. Bergman, J. Am. Chem. Soc., 1982, 104, 352 CrossRef.
- S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529 CrossRef.
- (a) E. C. Cherney, J. M. Lopchuk, J. C. Green and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 12592 CrossRef PubMed; (b) W. R. Gutekunst and P. S. Baran, J. Org. Chem., 2014, 79, 2430 CrossRef PubMed.
- D. S. Peters, F. E. Romesberg and P. S. Baran, J. Am. Chem. Soc., 2018, 140, 2072 CrossRef PubMed.
- H. M. L. Davies and D. Morton, J. Org. Chem., 2016, 81, 343 CrossRef PubMed.
- L. Ping, D. S. Chung, J. Bouffard and S.-G. Lee, Chem. Soc. Rev., 2017, 46, 4299 RSC.
- Y. Xu and G. Dong, Chem. Sci., 2018, 9, 1424 RSC.
- Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC.
- (a) Q. Bu, T. Rogge, V. Kotek and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 765 CrossRef PubMed; (b) R. Mei, S.-K. Zhang and L. Ackermann, Org. Lett., 2017, 19, 3171 CrossRef PubMed; (c) R. Mei, C. Zhu and L. Ackermann, Chem. Commun., 2016, 52, 13171 RSC; (d) J. Li and L. Ackermann, Chem. – Eur. J., 2015, 21, 5718 CrossRef PubMed; (e) S. Nakanowatari and L. Ackermann, Chem. – Eur. J., 2014, 20, 5409 CrossRef PubMed.
- S. De Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461 CrossRef.
- K. Ushiyama, N. Tanaka, H. Ono and H. Ogata, Jpn. J. Antibiot., 1971, 24, 197 CrossRef PubMed.
- (a) J. P. E. Human, A. W. Johnston, S. F. MacDonald and A. R. Todd, J. Chem. Soc., 1950, 477 RSC; (b) H. Duewell, A. W. Johnston, S. F. MacDonald and A. R. Todd, J. Chem. Soc., 1950, 485 RSC ; selected reviews on pyranonaphthoquinones: ; (c) M. A. Brimble, L. J. Duncalf and M. R. Nairna, Nat. Prod. Rep., 1999, 16, 267 RSC; (d) C. D. Donner, Nat. Prod. Rep., 2015, 32, 578 RSC.
- G. D'Andrea, Fitoterapia, 2015, 106, 256 CrossRef PubMed.
- K. Ingkaninan, A. P. Ijzerman and R. Verpoorte, J. Nat. Prod., 2000, 63, 315 CrossRef.
- (a) J. J. Nair, A. O. Aremu and J. van Staden, J. Ethnopharmacol., 2011, 137, 1102 CrossRef PubMed; (b) Z. Jin, Nat. Prod. Rep., 2013, 30, 849 RSC.
- (a) R. D. Appari, X. Chen, R. Chilukuri, A. P. Crew, H. Dong, C. Ferraro, K. Foreman, R. C. Gupta, A.-H. Li, D. Sherman, K. M. Stolz, B. Volk and R. Zahler, PCT Int. Appl., 2010141406, 2010 Search PubMed; (b) M. Kamata, T. Yamashita, T. Imaeda, T. Tanaka, S. Masada, M. Kamaura, S. Kasai, R. Hara, S. Sasaki, S. Takekawa, A. Asami, T. Kaisho, N. Suzuki, S. Ashina, H. Ogino, Y. Nakano, Y. Nagisa, K. Kato, K. Kato and Y. Ishihara, J. Med. Chem., 2012, 55, 2353 CrossRef PubMed.
- (a) E. N. da Silva Júnior, M. C. B. V. De Souza, A. V. Pinto, M. C. F. R. Pinto, M. O. F. Goulart, F. W. A. Barros, C. Pessoa, L. V. Costa-Lotufo, R. C. Montenegro, M. O. Moraes and V. F. Ferreira, Bioorg. Med. Chem., 2007, 15, 7035 CrossRef PubMed; (b) E. H. G. da Cruz, P. H. P. R. Carvalho, J. R. Corrêa, D. A. C. Silva, E. B. T. Diogo, J. D. de Souza Filho, B. C. Cavalcanti, C. Pessoa, H. C. B. de Oliveira, B. C. Guido, D. A. da Silva Filho, B. A. D. Neto and E. N. da Silva Júnior, New J. Chem., 2014, 38, 2569 RSC; (c) G. A. M. Jardim, T. T. Guimarães, M. C. F. R. Pinto, B. C. Cavalcanti, K. M. de Farias, C. Pessoa, C. C. Gatto, D. K. Nair, I. N. N. Namboothiri and E. N. da Silva Júnior, Med. Chem. Commun., 2015, 6, 120 RSC; (d) S. B. B. B. Bahia, W. J. Reis, G. A. M. Jardim, F. T. Souto, C. A. de Simone, C. C. Gatto, R. F. S. Menna-Barreto, S. L. de Castro, B. C. Cavalcanti, C. Pessoa, M. H. Araujo and E. N. da Silva Júnior, Med. Chem. Commun., 2016, 7, 1555 RSC; (e) B. C. Cavalcanti, F. W. A. Barros, I. O. Cabral, J. R. O. Ferreira, H. I. F. Magalhães, H. V. N. Júnior, E. N. da Silva Júnior, F. C. Abreu, C. O. Costa, M. O. F. Goulart, M. O. Moraes and C. Pessoa, Chem. Res. Toxicol., 2011, 24, 1560 CrossRef PubMed; (f) V. P. Andreev, E. G. Korvacheva and Ya. P. Nizhnik, Pharm. Chem. J., 2006, 40, 347 CrossRef; (g) P. R. Moore, G. J. Mannering, L. J. Teply and B. E. Kline, Cancer Res., 1960, 20, 628 Search PubMed.
- (a) K. S. Kanyiva, Y. Nakao and T. Hiyama, Angew. Chem., Int. Ed., 2007, 46, 8872 CrossRef PubMed; (b) S. H. Cho, S. J. Hwang and S. Chang, J. Am. Chem. Soc., 2008, 130, 9254 CrossRef PubMed; (c) J. Wu, X. Cui, L. Chen, G. Jiang and Y. Wu, J. Am. Chem. Soc., 2009, 131, 13888 CrossRef PubMed; (d) L.-C. Campeau, D. R. Stuart, J.-P. Leclerc, M. Bertrand-Laperle, E. Villemure, H.-Y. Sun, S. Lasserre, N. Guimond, M. Lecavallier and K. Fagnou, J. Am. Chem. Soc., 2009, 131, 3291 CrossRef PubMed; (e) Y. Araki, K. Kobayashi, M. Yonemoto and Y. Kondo, Org. Biomol. Chem., 2011, 9, 78 RSC; (f) J. Ryu, S. H. Cho and S. Chang, Angew. Chem., Int. Ed., 2012, 51, 3677 CrossRef PubMed; (g) Z. Y. Wu, C. Pi, X. L. Cui, J. Bai and Y. Wu, Adv. Synth. Catal., 2013, 355, 1971 CrossRef; (h) X. Chen, C. Zhu, X. Cui and Y. Wu, Chem. Commun., 2013, 49, 6900 RSC; (i) G. Li, C. Q. Jia and K. Sun, Org. Lett., 2013, 15, 5198 CrossRef PubMed.
- (a) X. Wang, Y. Ye, G. Ji, Y. Xu, S. Zhang, J. Feng, Y. Zhang and J. Wang, Org. Lett., 2013, 15, 3730 CrossRef PubMed; (b) J. Bian, X. Qian, N. Wang, T. Mu, X. Li, H. Sun, L. Zhang, Q. You and X. Zhang, Org. Lett., 2015, 17, 3410 CrossRef PubMed; (c) C. S. Lisboa, V. G. Santos, B. G. Vaz, N. C. de Lucas, M. N. Eberlin and S. J. Garden, J. Org. Chem., 2011, 76, 5264 CrossRef PubMed; (d) D. Wang, B. Ge, L. Li, J. Shan and Y. Ding, J. Org. Chem., 2014, 79, 8607 CrossRef PubMed; (e) A. Arnone, L. Merlini, G. Nasini and O. V. de Pava, Synth. Commun., 2007, 37, 2569 CrossRef; (f) G. Naturale, M. Lamblin, C. Commandeur, F.-X. Felpin and J. Dessolin, Eur. J. Org. Chem., 2012, 5774 CrossRef.
- (a) J. P. Michael, Nat. Prod. Rep., 2008, 25, 166 RSC; (b) A. B. Burgin, O. T. Magnusson, J. Singh, P. Witte, B. L. Staker, J. M. Bjornsson, M. Thorsteinsdottir, S. Hrafnsdottir, T. Hagen, A. S. Kiselyov, L. J. Stewart and M. E. Gurney, Nat. Biotechnol., 2010, 28, 63 CrossRef PubMed; (c) T. Eicher and S. Hauptmann, The Chemistry of Heterocycles, WIleyVCH, Weinheim, 2nd edn 2003 CrossRef.
- J. Ryu, S. H. Cho and S. Chang, Angew. Chem., Int. Ed., 2012, 51, 3677 CrossRef PubMed.
- (a) D.-Y. Li, Z.-L. Huang and P.-N. Liu, Org. Lett., 2018, 20, 2028 CrossRef PubMed; (b) X. Chen, X. Cui and Y. Wu, Org. Lett., 2016, 18, 3722 CrossRef PubMed; (c) R. Sharma, R. Kumar, I. Kumar and U. Sharma, Eur. J. Org. Chem., 2015, 7519 CrossRef; (d) U. Sharma, Y. Park and S. Chang, J. Org. Chem., 2014, 79, 9899 CrossRef PubMed; (e) J. Jeong, P. Patel, H. Hwang and S. Chang, Org. Lett., 2014, 16, 4598 CrossRef PubMed; (f) J. Kwak, Y. Ohk, Y. Jung and S. Chang, J. Am. Chem. Soc., 2012, 134, 17778 CrossRef PubMed; (g) Y. Shang, X. Jie, H. Zhao, P. Hu and W. Su, Org. Lett., 2014, 16, 416 CrossRef PubMed.
- (a) V. M. Dembitsky, T. A. Gloriozova and V. V. Poroikov, Phytomedicine, 2015, 22, 183 CrossRef PubMed; (b) O. M. Yaghi, H. Li, C. Davis, D. Richardson and T. L. Groy, Acc. Chem. Res., 1998, 31, 474 CrossRef; (c) H. Usta, A. Facchetti and T. J. Marks, Acc. Chem. Res., 2011, 44, 501 CrossRef PubMed.
- (a) L.-C. Campeau, M. Bertrand-Laperle, J.-P. Leclerc, E. Villemure, S. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 3276 CrossRef PubMed; (b) F. Roudesly, J. Oble and G. Poli, J. Mol. Catal. A: Chem., 2017, 426, 275 CrossRef; (c) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787 CrossRef PubMed; (d) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef PubMed ; for detailed computational studies on the position-selectivity, see: ; (e) C. Deng, W. H. Lam and Z. Lin, Organometallics, 2017, 36, 650 CrossRef; (f) X. Yilu, Y. Shengwen, D. Lijuan and L. Juan, Eur. J. Org. Chem., 2017, 381 Search PubMed; (g) J.-B. Liu, X.-H. Sheng, C.-Z. Sun, F. Huang and D.-Z. Chen, ACS Catal., 2016, 6, 2452 CrossRef.
- D. E. Stephens, J. Lakey-Beitia, A. C. Atesin, T. A. Atesin, G. Chavez, H. D. Arman and O. V. Larionov, ACS Catal., 2015, 5, 167 CrossRef PubMed.
- D. E. Stephens, J. Lakey-Beitia, G. Chavez, C. Ilie, H. D. Arman and O. V. Larionov, Chem. Commun., 2015, 51, 9507 RSC.
- T. Shibata and Y. Matsuo, Adv. Synth. Catal., 2014, 356, 1516 CrossRef.
- H. Hwang, J. Kim, J. Jeong and S. Chang, J. Am. Chem. Soc., 2014, 136, 10770 CrossRef PubMed.
- K. Shin, S.-W. Park and S. Chang, J. Am. Chem. Soc., 2015, 137, 8584 CrossRef PubMed.
- D. Kalsi, R. A. Laskar, N. Barsu, J. R. Premkumar and B. Sundararaju, Org. Lett., 2016, 18, 4198 CrossRef PubMed.
- N. Barsu, M. Sen, J. R. Premkumar and B. Sundararaju, Chem. Commun., 2016, 52, 1338 RSC.
- B. Wang, C. Li and H. Liu, Adv. Synth. Catal., 2017, 359, 3029 CrossRef.
- K. Kitazawa, T. Kochi, M. Sato and F. Kakiuchi, Org. Lett., 2009, 11, 1951 CrossRef PubMed.
- D. Matsumura, K. Kitazawa, S. Terai, T. Kochi, Y. Le, M. Nitani, Y. Aso and F. Kakiuchi, Org. Lett., 2012, 14, 3882 CrossRef PubMed.
- M. Wang, C. Zhang, L.-P. Sun, C. Ding and A. Zhang, J. Org. Chem., 2014, 79, 4553 CrossRef PubMed.
- C. Zhang, M. Wang, Z. Fan, L.-P. Sun and A. Zhang, J. Org. Chem., 2014, 79, 7626 CrossRef PubMed.
- G. A. M. Jardim, E. N. da Silva Júnior and J. F. Bower, Chem. Sci., 2016, 7, 3780 RSC.
- G. A. M. Jardim, T. L. Silva, M. O. F. Goulart, C. A. de Simone, J. M. C. Barbosa, K. Salomão, S. L. de Castro, J. F. Bower and E. N. da Silva Júnior, Eur. J. Med. Chem., 2017, 136, 406 CrossRef PubMed.
- G. A. M. Jardim, W. X. C. Oliveira, R. P. de Freitas, R. F. S. Menna-Barreto, T. L. Silva, M. O. F. Goulart and E. N. da Silva Júnior, Org. Biomol. Chem., 2018, 16, 1686 Search PubMed.
- J. Wang, D. Qin, J. Lan, Y. Cheng, S. Zhang, Q. Guo, J. Wu, D. Wu and J. You, Chem. Commun., 2015, 51, 6337 RSC.
- D. Kang and S. Hong, Org. Lett., 2015, 17, 1938 CrossRef PubMed.
- A. C. Shaikh, D. R. Shinde and N. T. Patil, Org. Lett., 2016, 18, 1056 CrossRef PubMed.
- A. Banerjee, S. K. Santra, P. R. Mohanta and B. K. Patel, Org. Lett., 2015, 17, 5678 CrossRef PubMed.
- M. P. Huestis, J. Org. Chem., 2016, 81, 12545 CrossRef PubMed.
- S. H. Han, S. Kim, U. De, N. K. Mishra, J. Park, S. Sharma, J. H. Kwak, S. Han, H. S. Kim and I. S. Kim, J. Org. Chem., 2016, 81, 12416 CrossRef PubMed.
- K. Kim, H. Choe, Y. Jeong, J. H. Lee and S. Hong, Org. Lett., 2015, 17, 2550 CrossRef PubMed.
- (a) Y. Yang, Y. Lin and Y. Rao, Org. Lett., 2012, 14, 2874 CrossRef PubMed; (b) V. S. Thirunavukkarasu and L. Ackermann, Org. Lett., 2012, 14, 4210 CrossRef PubMed; (c) V. S. Thirunavukkarasu, J. Hubrich and L. Ackermann, Org. Lett., 2012, 14, 6206 CrossRef PubMed; (d) F. Yang and L. Ackermann, Org. Lett., 2013, 15, 718 CrossRef PubMed; (e) W. Liu and L. Ackermann, Org. Lett., 2013, 15, 3484 CrossRef PubMed; (f) F. Yang, K. Rauch, K. Kettelhoit and L. Ackermann, Angew. Chem., Int. Ed., 2014, 53, 11285 CrossRef PubMed.
- R. Samanta, R. Narayan and A. P. Antonchick, Org. Lett., 2012, 14, 6108 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |