
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Methyl-rotation dynamics in metal–organic frameworks probed with terahertz spectroscopy†
Qi
Li
 a,
Adam J.
Zaczek
b,
Timothy M.
Korter
b,
J. Axel
Zeitler
a and
Michael T.
Ruggiero
*ac
a,
Adam J.
Zaczek
b,
Timothy M.
Korter
b,
J. Axel
Zeitler
a and
Michael T.
Ruggiero
*ac
aDepartment of Chemical Engineering and Biotechnology, University of Cambridge, Philippa Fawcett Drive, Cambridge, CB3 0AS, UK
bDepartment of Chemistry, Syracuse University, 1-014 Center for Science and Technology, Syracuse, NY 13244, USA
cDepartment of Chemistry, University of Vermont, 82 University Place, Burlington, VT 05405, USA. E-mail: mtr34@cam.ac.uk
First published on 14th May 2018
Abstract
In ZIF-8 and its cobalt analogue ZIF-67, the imidazolate methyl-groups, which point directly into the void space, have been shown to freely rotate – even down to cryogenic temperatures. Using a combination of experimental terahertz time-domain spectroscopy, low-frequency Raman spectroscopy, and state-of-the-art ab initio simulations, the methyl-rotor dynamics in ZIF-8 and ZIF-67 are fully characterized within the context of a quantum-mechanical hindered-rotor model. The results lend insight into the fundamental origins of the experimentally observed methyl-rotor dynamics, and provide valuable insight into the nature of the weak interactions present within this important class of materials.
Metal–organic frameworks (MOFs) are a class of solid materials that have exciting physical properties with numerous applications including gas storage, chemical separation, drug delivery, and catalysis.1–5 The basis of the diverse utility of MOFs partly lies in their rich topological possibilities.6,7 A wide range of three-dimensional structures can be realized, with many of these containing large spatial voids that are related to their applicability.8–10 Unquestionably, the molecular and bulk structures of MOFs play a major role in defining their performance for such applications.11–13 It has however become clear in recent years that the vibrational dynamics of the solids are also of critical importance in dictating the efficacy of such materials.14–16 Previous work has demonstrated that in particular, the low-frequency (sub 200 cm−1) vibrational motions are clearly related to many properties, such as mechanochemical and gas adsorption phenomena.17,18 It is therefore highly desirable to develop a complete understanding of the interplay between the structural and dynamical aspects of MOFs in order to optimize their design, and ultimately, their function.
In one of the most well-studied MOF systems, ZIF-8, it has been shown that the solid is capable of absorbing a large variety of molecules, ranging from gaseous nitrogen to active pharmaceutical ingredients.19–21 In ZIF-8 and its cobalt-substituted analogue, ZIF-67,22,23 the methyl-groups on the imidazolate linkers that point into the pore exhibit free (or nearly-free) rotation, giving rise to very low-energy transitions on the order of μeV.24 This class of quasi-hindered-rotational motions have long been used as a probe of the weak forces within materials, and thus is an important aspect to consider in these two materials.25–27 By building on the existing understanding of the fundamental quantum mechanical processes underpinning these motions, we aim to develop better insight into the properties of ZIF-8 and ZIF-67.
Low-frequency vibrational spectroscopy (10–200 cm−1 or 1–25 meV) provides unique insight into the weak forces present within molecular solids.28–30 Since the vibrational motions often involve large amplitude motions of entire molecules, it is possible to experimentally explore a large portion of both the intra- and intermolecular potential energy hypersurface.31 By combining the experimental data with quantum mechanical simulations, it is possible to fully characterize the atomic-level structure, dynamics, and forces that are present within the solid.32–35 Because of the inherent dependence on a rigorous description of the weak forces, the accurate simulation of the low-frequency vibrational spectra provides a benchmark of the theoretical approach.30,36 A good match between the experimental vibrational spectrum and the simulated spectra can be considered a validation method for the ab initio simulations, enabling additional data that would be difficult to obtain experimentally (e.g. band structures), to be extracted with a high degree of confidence. In this work, experimental terahertz time-domain spectroscopy (THz-TDS) as well as low-frequency Raman spectroscopy are combined with solid-state density functional theory (DFT) simulations in order to probe the quantum mechanical origin and atomic dynamics of the quasi-free methyl rotation in crystals of ZIF-8 and ZIF-67.
In order to understand these phenomena, both solids were investigated using fully-periodic DFT calculations with the Crystal17 software package.37 The M06-2X meta-GGA density functional was used for correlation and exchange (with 54% explicit Hartree–Fock exchange),38 and the split-valence double-ζ 6-31G(d,p) basis set39 was utilized for all calculations (additional methodology details available in the ESI†). The simulated structures of ZIF-8 and ZIF-67 were in excellent agreement with the experimental data. For ZIF-8, the calculation yielded an error in the lattice parameter of 0.3% compared to the published structure, and root-mean-squared deviations (RMSDs) of bond length and angle of 0.04 Å, and 3.20°, respectively. The simulation of ZIF-67 was equally as accurate, with a lattice parameter error of 0.6%, and RMSDs of the bonds and angles of 0.02 Å, and 1.12°, respectively. All further methyl-rotation calculations were based on these predicted structures. A comparison of the two topologies shows that there is very little deviation in internal conformation of the imdizolate linkers, with most bonds predicted to have the same (or very similar) lengths and angles, while the ZIF-67 crystal packing is slightly contracted compared to that of ZIF-8 (−1.5% contraction in unit cell volume).
The similarities in the structures translates to equally similar low-frequency vibrational spectra. The experimental THz-TDS and low-frequency Raman spectra of the two materials (Fig. 1) show a number of well-resolved absorption features by both methods. Only minor shifting (<5 cm−1) is observed for a few absorption features.
 | ||
| Fig. 1 The experimental low-frequency vibrational spectra of ZIF-8 (blue) and ZIF-67 (red). The left panel shows the 78 K THz-TDS spectra, as well as the simulated IR-spectra (black) convolved with Lorentzian line-shapes based on the experimental full-width at half-maxima, and corresponding vibrational transitions (black sticks). The right panel shows the experimental 298 K low-frequency Raman spectra, with the M06-2X-predicted Raman-active modes and PBE-predicted spectra shown as solid and dashed lines, respectively. | ||
In order to interpret the low-frequency spectra, vibrational analyses were performed using Crystal17 using a two-point numerical differentiation scheme. The results of the calculations, also shown in Fig. 1, yield a good agreement with the experimental data. With respect to the THz-TDS data, the experimental frequencies and intensities are well-reproduced by the theoretical model, with a slight over-estimation of the intensity of the modes occurring experimentally near 130 cm−1 in both solids. Because the simulation of Raman intensities using M06-2X is not supported by the crystal code, Raman intensities were simulated using the PBE functional.40 It is important to note that with PBE there exists a negative vibrational mode corresponding to the methyl rotation that results purely from numerical inaccuracies, originating from fitting the extremely shallow (but positive) potential with a harmonic oscillator. However, the results are in generally good agreement with both the experimental spectra and the M06-2X predicted transitions.
Investigation of the vibrational normal modes highlights that the motions associated with these modes are large-amplitude displacements of the atoms within the solid, primarily with respect to the imidazolate linkers. Of particular interest in the context of this study are vibrations that involve significant methyl-motion, which is present in many of the low-frequency modes that are both IR- and Raman-active. The modes that occur between 120–140 cm−1 primarily involve hindered-rotational motion. It is important to note that none of these motions represent pure hindered-rotational motion, but are rather complex mode-types that involve simultaneous torsions or hindered-rotations of the imidazolate linkers as well. Nevertheless, the successful modeling of the experimental low-frequency spectra lends significant confidence that the theoretical model is accurately reproducing the weak forces present within these solids, including the methyl-rotational potential energy surface, and permits deeper investigation.
In order to explore the hindered-rotational motion of the imidazolate methyl-groups further, the potential energy curve related to that coordinate was explicitly determined by performing a relaxed scan of the methyl-rotation angle in steps of 10°. The results of these calculations, shown in Fig. 2, show that the two potentials are similar and are rather shallow in both cases. The potentials were found to contain three-fold symmetry, and as such only the 0–120° domain is shown. Both solids display the same general shape, with the ZIF-67 potential slightly stiffer than that of ZIF-8. It should be noted that these energies represent calculations performed with full crystalline symmetry and periodic boundary conditions, are given in units of meV mol−1 methyl−1. The role of coupled motion was explored further by removing symmetry and rotating a single methyl-unit in the ZIF-8 crystal. The calculations yielded nearly identical potentials, indicating that the individual methyl groups are not interacting significantly, in good agreement with experimental accounts.24
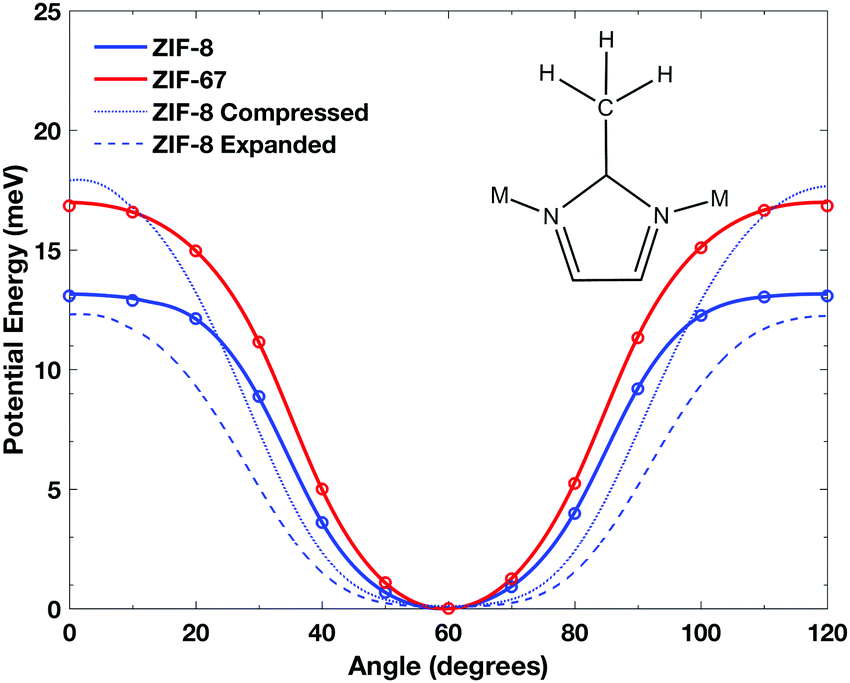 | ||
| Fig. 2 Simulated methyl rotor potential energy of ZIF-8 (blue) and ZIF-67 (red) with a step of 10° from 0° to 120° with the 60° minimum representing the equilibrium staggered conformation of the methyl-hydrogens with respect to the plane of the imidazolate ring. The blue dotted and dashed lines represent a contracted and expanded ZIF-8 structure, respectively. | ||
The explicit determination of the periodic methyl-rotation potential makes it possible to explore the fundamental basis of the observed dynamics. Based on the rotational potential energy, the explicit vibrational energy levels were determined in the absence of the harmonic approximation, as well as the corresponding vibrational wavefunctions, by solving the one-dimensional Schrödinger equation as suggested by Lewis et al.41 The results, shown in Fig. 3, highlights the complex nature of the hindered-rotational dynamics in these solids. The data are in agreement with similar systems,42 where quantum tunneling is known to result in a splitting of the triply degenerate ground state into two discrete levels.43
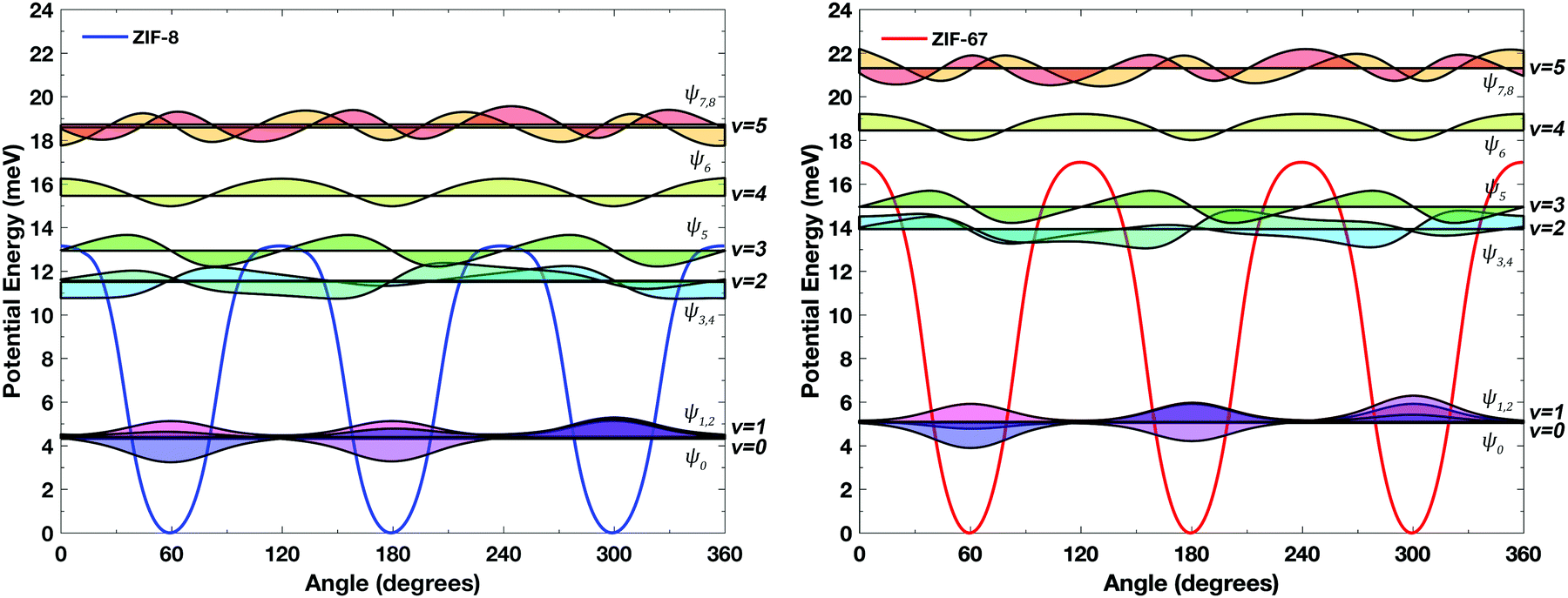 | ||
| Fig. 3 Simulated methyl rotor potential energy of ZIF-8 (left, blue) and ZIF-67 (right, red) with the torsional vibrational levels drawn as black lines, and the scaled-normalized (×3) vibrational wavefunctions drawn as shaded curves. | ||
One of the major considerations is the rotational barrier, which was previously determined experimentally to be ≈6–8 meV from inelastic neutron scattering experiments performed on ZIF-8.24 While the barrier that is predicted for ZIF-8 is seemingly larger (13.16 meV), it is important to take zero-point energy into consideration. In doing so, the resulting barrier is much closer to the experimental data, yielding values of 8.85 meV and 11.95 meV for ZIF-8 and ZIF-67, respectively. These energies correspond to an equivalent temperature of 102.7 K and 138.7 K, which explains the observed free rotation at cryogenic temperatures. The higher-energy vibrational levels exhibit characteristics of a plane wave commensurate with the lack of any potential energy bounds. This result indicates that free rotation is possible and indeed likely. Furthermore, if the temperature was reduced sufficiently as to trap the rotor in the potential well, this would still not completely remove large-amplitude rotations as the lowest-lying vibrational wavefunctions (shaded purple in Fig. 3) show significant probability in the classically forbidden region. This is further evidence in support of previous literature suggestions of methyl tunneling, and matches with the large thermal ellipsoids obtained from neutron diffraction experiments at 3.5 K.24
The deviations between the two ZIF systems is somewhat surprising, given that the structures and vibrational dynamics are so similar. However, it is important to note that the ZIF-67 crystal is slightly more contracted than ZIF-8, which appears to be sufficient enough to increase the barrier to rotation as observed. Additionally, a minor deviation between ZIF-8 and ZIF-67 in the internal bond angle between the imidazole ring and the methyl-carbon is observed that increases from 0.15–0.25 with the same trend as the deviations in the two potentials as a function of angle. While this is clearly very subtle, such phenomena might be significant to altering the rotor-potential in a ‘through-bond’ manner, which has been observed previously.44 In order to achieve a better understanding, simulations were performed on ZIF-8 where the volume was chosen to match ZIF-67 (1.5% contraction), as well as expanding the volume by 1.5%, and the results are shown in Fig. 2. While the compression ultimately results in a higher potential barrier (with the inverse true for expansion), both scenarios result in a more shallow potential curve at small displacements. This can be rationalized within the through-bond picture, as changes in the covalent bonds from the changing lattice vector ultimately result in a weaker carbon–carbon bond, softening the rotational potential.
Closer analysis reveals that the simulations are able to provide additional insight into the previously reported experimental neutron scattering data. The predicted energy of the fundamental hindered-rotational transition predicted at 224 μeV is in good agreement with the value reported by Zhou et al. of 334 μeV (see Table 1). The 1 → 2 transition, reported to occur at 2.7 meV, seemingly disagrees with the simulated value. However, the predicted 1 → 2 energy is outside the range of the instrument used for the experimental neutron measurements, and based on the results of the simulation it is more likely that the experimentally observed feature is actually the 2 → 3 transition, which is predicted occur at 1.43 meV in ZIF-8. It is important to note that while the simulations generally overestimate the experimental results, the errors in the absolute values correspond to very small energy deviations. For example, an error of 100 μeV (2.3 × 10−3 kcal mol−1) is three orders of magnitude lower than the established “chemical accuracy” criterion of 1 kcal mol−1.45,46 Overall, the values obtained highlight the substantial accuracy of the quantum mechanical simulations, and indicate that even the weakest forces within these solids are very well-described by the simulations.
QL, JAZ, and MTR thank the UK Engineering and Physical Sciences Research Council (EPSRC) for funding (EP/N022769/1) and for access to the ARCHER UK national supercomputer (EP/L000202). The authors also thank Toptica Photonics for use of their TeraFlash THz-TDS system.
| Experimental24 | Simulated | ||
|---|---|---|---|
| ZIF-8 | ZIF-8 | ZIF-67 | |
| Potential max. | — | 13.16 | 16.99 |
| Zero-point | — | 4.31 | 5.04 |
| Barrier height | ∼6–8 | 8.85 | 11.95 |
| Transition energies | |||
|---|---|---|---|
| ZIF-8 | ZIF-8 | ZIF-67 | |
| 0 → 1 | 334 × 10−3 | 225 × 10−3 | 466 × 10−3 |
| 1 → 2 | 2.7 | 6.97 | 8.83 |
| 2 → 3 | — | 1.43 | 1.02 |
Conflicts of interest
There are no conflicts to declare.References
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef PubMed.
- B. R. Pimentel, A. Parulkar, E. K. Zhou, N. A. Brunelli and R. P. Lively, ChemSusChem, 2014, 7, 3202–3240 CrossRef PubMed.
- M. H. Teplensky, M. Fantham, P. Li, T. C. Wang, J. P. Mehta, L. J. Young, P. Z. Moghadam, J. T. Hupp, O. K. Farha, C. F. Kaminski and D. Fairen-Jimenez, J. Am. Chem. Soc., 2017, 139, 7522–7532 CrossRef PubMed.
- D. T. Sun, L. Peng, W. S. Reeder, S. M. Moosavi, D. Tiana, D. K. Britt, E. Oveisi and W. L. Queen, ACS Cent. Sci., 2018, 4, 349–356 CrossRef PubMed.
- B. Chen, Z. Yang, Y. Zhu and Y. Xia, J. Mater. Chem. A, 2014, 2, 16811–16831 Search PubMed.
- a. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- P. Z. Moghadam, A. Li, S. B. Wiggin, A. Tao, A. G. P. Maloney, P. A. Wood, S. C. Ward and D. Fairen-Jimenez, Chem. Mater., 2017, 29, 2618–2625 CrossRef.
- B. J. Sikora, R. Winnegar, D. M. Proserpio and R. Q. Snurr, Microporous Mesoporous Mater., 2014, 186, 207–213 CrossRef.
- J. T. A. Jones, T. Hasell, X. Wu, J. Bacsa, K. E. Jelfs, M. Schmidtmann, S. Y. Chong, D. J. Adams, A. Trewin, F. Schiffman, F. Cora, B. Slater, A. Steiner, G. M. Day and A. I. Cooper, Nat. Commun., 2011, 474, 367–371 CrossRef PubMed.
- D. Nazarian, J. S. Camp and D. S. Sholl, Chem. Mater., 2016, 28, 785–793 CrossRef.
- A. U. Ortiz, A. Boutin, A. H. Fuchs and F.-X. Coudert, J. Phys. Chem. Lett., 2013, 4, 1861–1865 CrossRef PubMed.
- M. Witman, S. Ling, S. Jawahery, P. G. Boyd, M. Haranczyk, B. Slater and B. Smit, J. Am. Chem. Soc., 2017, 139, 5547–5557 CrossRef PubMed.
- X. Wang, R. Guo, D. Xu, J. Chung, M. Kaviany and B. Huang, J. Phys. Chem. C, 2015, 119, 26000–26008 Search PubMed.
- N. Lock, Y. Wu, M. Christensen, L. J. Cameron, V. K. Peterson, A. J. Bridgeman, C. J. Kepert and B. B. Iversen, J. Phys. Chem. C, 2010, 114, 16181–16186 Search PubMed.
- F.-X. Coudert, ChemPhysChem, 2017, 18, 2732–2738 CrossRef PubMed.
- T. Tanno, Y. Watanabe, K. Umeno, A. Matsuoka, H. Matsumura, M. Odaka and N. Ogawa, J. Phys. Chem. C, 2017, 121, 17921–17924 Search PubMed.
- W. Zhang, J. Maul, D. Vulpe, P. Z. Moghadam, D. Fairen-Jimenez, D. M. Mittleman, J. A. Zeitler, A. Erba and M. T. Ruggiero, ChemRxiv DOI:10.26434/chemrxiv.6244892.v1.
- D. Fairen-Jimenez, S. A. Moggach, M. T. Wharmby, P. A. Wright, S. Parsons and T. Düren, J. Am. Chem. Soc., 2011, 133, 8900–8902 CrossRef PubMed.
- L. Mu, B. Liu, H. Liu, Y. Yang, C. Sun and G. Chen, J. Mater. Chem., 2012, 22, 12246–12252 RSC.
- I. B. Vasconcelos, T. G. Da Silva, G. C. G. Militão, T. A. Soares, N. M. Rodrigues, M. O. Rodrigues, N. B. Da Costa, R. O. Freire and S. A. Junior, RSC Adv., 2012, 2, 9437–9442 RSC.
- J. Qian, F. Sun and L. Qin, Mater. Lett., 2012, 82, 220–223 CrossRef.
- Q. Shi, Z. Chen, Z. Song, J. Li and J. Dong, Angew. Chem., Int. Ed., 2011, 50, 672–675 CrossRef PubMed.
- W. Zhou, H. Wu, T. J. Udovic, J. J. Rush and T. Yildirim, J. Phys. Chem. A, 2008, 112, 12602–12606 CrossRef PubMed.
- R. R. Lozada Garcia, J. Ceponkus, M. Chevalier, W. Chin, J. M. Mestdagh and C. Crépin, Angew. Chem., Int. Ed., 2012, 51, 6947–6950 CrossRef PubMed.
- J. Assmann, R. V. Benten, A. Charvat and B. Abel, J. Phys. Chem. A, 2003, 107, 1904–1913 CrossRef.
- D. B. Moss and C. S. Parmenter, J. Chem. Phys., 1998, 98, 6897–6905 CrossRef.
- E. P. J. Parrott and J. A. Zeitler, Appl. Spectrosc., 2015, 69, 1–25 CrossRef PubMed.
- M. T. Ruggiero and T. M. Korter, Phys. Chem. Chem. Phys., 2016, 18, 5521–5528 RSC.
- M. T. Ruggiero, J. A. Zeitler and A. Erba, Chem. Commun., 2017, 53, 3781–3784 RSC.
- M. T. Ruggiero, M. Krynski, E. O. Kissi, J. Sibik, D. Markl, N. Y. Tan, D. Arslanov, W. Van Der Zande, B. Redlich, T. M. Korter, H. Grohganz, K. Löbmann, T. Rades, S. R. Elliott and J. A. Zeitler, Phys. Chem. Chem. Phys., 2017, 19, 30039–30047 RSC.
- A. J. Zaczek and T. M. Korter, Cryst. Growth Des., 2017, 17, 4458–4466 Search PubMed.
- M. R. C. Williams, D. J. Aschaffenburg, B. K. Ofori-Okai and C. A. Schmuttenmaer, J. Phys. Chem. B, 2013, 117, 10444–10461 CrossRef PubMed.
- M. T. Ruggiero, J. Sibik, J. A. Zeitler and T. M. Korter, J. Phys. Chem. A, 2016, 120, 7490 CrossRef PubMed.
- M. T. Ruggiero, J. Sibik, A. Erba, J. A. Zeitler and T. M. Korter, Phys. Chem. Chem. Phys., 2017, 19, 28647–28652 RSC.
- G. J. O. Beran, Chem. Rev., 2016, 116, 5567–5613 CrossRef PubMed.
- R. Dovesi, A. Erba, R. Orlando, C. M. Zicovich Wilson, B. Civalleri, L. Maschio, M. Rérat, S. Casassa, J. Baima, S. Salustro and B. Kirtman, WIREs Comput. Mol. Sci., 2018, 110, e1360 CrossRef.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120, 215–241 CrossRef.
- P. C. Hariharan and J. A. Pople, Mol. Phys., 2006, 27, 209–214 CrossRef.
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef PubMed.
- J. D. Lewis, T. B. Malloy Jr., T. H. Chao and J. Laane, J. Mol. Struct., 1972, 12, 427–449 CrossRef.
- D. R. Herschbach, J. Chem. Phys., 1959, 31, 91–108 CrossRef.
- S. Khazaei and D. Sebastiani, J. Chem. Phys., 2017, 147, 194303 CrossRef PubMed.
- W. J. Hehre, J. A. Pople and A. J. P. Devaquet, J. Am. Chem. Soc., 1976, 98, 664–668 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc02650e |
| This journal is © The Royal Society of Chemistry 2018 |