
Self-doped colloidal semiconductor nanocrystals with intraband transitions in steady state
Jihye
Kim†
 ,
Dongsun
Choi†
and
Kwang Seob
Jeong
*
,
Dongsun
Choi†
and
Kwang Seob
Jeong
*
Department of Chemistry, Korea University, Seoul 02841, Republic of Korea. E-mail: kwangsjeong@korea.ac.kr
First published on 26th June 2018
Abstract
The tunable bandgap energy has been recognized as a prominent feature of the colloidal semiconductor nanocrystal, also called the colloidal quantum dot (CQD). Due to the broken degeneracy caused by the quantum confinement effect, the electronic states of the conduction band (CB) are separated by a few hundred meV. The electronic transition occurring in the conduction band is called the intraband transition and has been regarded as a fast electron relaxation process that cannot be readily observed under steady state. However, recent progress in the studies of intraband transitions allowed the observation of the mid-IR intraband transition in steady state and ambient condition, providing a pathway to exploit the mid-IR electronic transition for various optoelectronic applications. The observation of the steady state intraband transitions has been possible due to the electron filling of the lowest electronic state (1Se) of the conduction band in the semiconductor nanocrystal. Specifically, the nanocrystals are “self-doped” with electrons through chemical synthesis – that is, without the need of adding heterogeneous impurity or applying an electrical potential. In this feature article, we summarize the recent advances in the study on intraband electronic transitions along with the interesting findings on the magnetic and electronic properties of the self-doped colloidal metal chalcogenide semiconductor nanocrystals. The mid-IR intraband transitions of non-toxic nanocrystals, which exclude the toxic mercury and cadmium constituents, are also highlighted, which hold promise for safer applications utilizing the higher quantum states of nanocrystals.
 Jihye Kim | Jihye Kim earned a BS in chemistry from Korea University and a PhD in chemistry from the Pennsylvania State University (Advisor: Professor John Asbury) focusing on transient infrared spectroscopy to investigate charge transport in colloidal semiconductor nanocrystals. She is currently a lecturer in the Department of Chemistry at Korea University. |
 Dongsun Choi | Dongsun Choi is pursuing a PhD in chemistry at Korea University under the supervision of Professor Kwang Seob Jeong. He earned his BS in chemistry from the University of Colorado at Boulder in 2015. His current research topics focus on the synthesis of mid-infrared intraband producing quantum dots and optical studies to examine the properties of intraband transitions. |
 Kwang Seob Jeong | Kwang Seob Jeong is an assistant professor in the Department of Chemistry at Korea University in Seoul, Korea. He obtained his PhD in chemistry from the Pennsylvania State University (Advisor: Professor John Asbury), followed by post-doctoral training at the University of Chicago with Professor Philippe Guyot-Sionnest. He started his independent career at Korea University in 2015. His main research focuses on exploring electronic transitions in colloidal nanomaterials. |
Introduction
The colloidal quantum dot (CQD) has been of great interest in bioimaging,1,2 electronic,3,4 optoelectronic5,6 and photovoltaic applications7,8 due to a variety of advantages such as the size-tunable bandgap transition,9,10 the solution-based synthesis process and the availability of low-temperature manufacturing.6,11–14 For more than three decades, continuous studies on CQDs have made significant improvement, and nowadays one can readily find commercial electronic devices equipped with colloidal quantum dot displays.Upon photoexcitation, an electron in the valence band of the semiconductor nanocrystal is excited to the electronic states of the conduction band (CB). The electron in the higher electronic states of the CB, called a hot-electron, is known to immediately relax to the band edge, or the lowest electronic state of the conduction band (1Se). In this process, the electron can interact with phonons (crystal vibration modes).15 Then, the electron at 1Se recombines with a hole in the valence band, resulting in the photoluminescence (PL, radiative recombination). Therefore, it is essential to understand this hot-electron relaxation process that takes place before the bandgap recombination process to fully understand all the possible carrier kinetic processes triggered by the photoexcitation, which eventually determine the performance of the optoelectronic applications.3–8,16
The intraband transition refers to the transition occurring between the discrete electronic states such as the 1Se–1Pe transition. Therefore, the study on intraband transition will provide valuable information on the hot-carrier dynamics. The intraband transition is also important in utilizing a wider optical range. The intraband transition is the only way to use the energy smaller than the constant bulk bandgap energy without changing the material constituent. This is because the nanocrystal bandgap energy (Eg,NC) is a sum of the bulk bandgap (Eg,bulk) and quantum confinement effect, where the quantum confinement effect is always positive, and thus the nanocrystal bandgap energy is always greater than the bulk bandgap.
For example, the bulk bandgap energy of β-HgS is 0.65 eV and the intraband transition energy is ∼0.12–0.4 eV, which enables the use of energy smaller than the constant bulk β-HgS bandgap energy.
In addition, the intraband transition is also tunable by varying the nanocrystal size since the origin of the transition is the quantum confinement effect. It produces a distinct Gaussian peak whereas the bandgap transition is typically overlapped with other interband transition. In other words, the intraband transition is superior to the bandgap transition in terms of wavelength selectivity. One thing to note is that the clear optical peak of the intraband transition is only observed in the three-dimensionally quantum confined colloidal quantum dots while quantum wires or quantum wells cannot give distinct intraband transition peaks.
The advantage of the colloidal nanocrystal is the transition energy tunability by merely varying the size of the nanocrystal. Due to the accessibility to such small energy, the intraband transition allows the colloidal nanocrystals to dramatically expand the accessible energy range from UV to THz radiation.
Several experimental methods have been carried out to examine the intraband transitions of colloidal nanocrystals. The powerful method to study the carrier dynamics is the transient spectroscopy to scrutinize the carrier relaxation as a function of time. However, the native fast carrier relaxation of the hot electron cannot guarantee the virtual use of the intraband transitions, and thereby, alternative methods were introduced by several groups. The key concept of these alternative methods is to incorporate excess carriers into the nanocrystals.
The addition of excess carriers in the nanocrystal can be achieved by several methods that have been carefully investigated for the last two decades.16 Charge transfer (or chemical) doping,17 electrochemical doping,19 photochemical doping,20 and self-doping21 methods are the examples.
The injection of the carriers into the nanocrystals with discrete energy levels make it possible to observe the intraband transition in the infrared regime corresponding to the transition from the lowest electronic state (1Se) to the second lowest electronic state (1Pe).22–30 The energy of the intraband transition occurring between 1Se–1Pe is ∼0.1–0.5 eV which is larger than the longitudinal optical (LO) phonon by a factor of 3 to 20 depending on the composition and the size of the nanocrystal. For instance, the intraband transition of the HgS CQD is ∼0.12–0.4 eV and its LO phonon is 0.031 eV.24 Thus the less effective coupling between the intraband transition and the LO phonon enables a clear observation of the intraband transition.
This feature article focuses on the methods that have been used for observing the intraband transition of colloidal nanocrystals and provides future direction of the study of the electronic transitions occurring in the higher quantum states of colloidal nanocrystals.
Ultrafast spectroscopy
The study of the intraband transition was initiated during the end of the last century. Guyot-Sionnest and his co-workers first started the intraband transition of colloidal nanocrystal study by performing the picosecond visible pump infrared probe spectroscopy to directly measure the transient intraband absorption of CdSe colloidal quantum dots in the mid-IR regime in 1998 (Fig. 1A and B).22,25 Klimov and his co-workers also carried out the visible electronic pump–probe spectroscopy to measure the interband exciton relaxation while the intraband transition could indirectly calculated (Fig. 1C).26 Since then, the time-resolved intraband spectroscopy for the CQDs has been intensively performed to understand the hot carrier dynamics. Due to the coulomb potential between the electron and hole, the intraband transition is extremely fast, so that the carrier dynamic information can be obtained merely by ultrafast spectroscopy (Fig. 1D).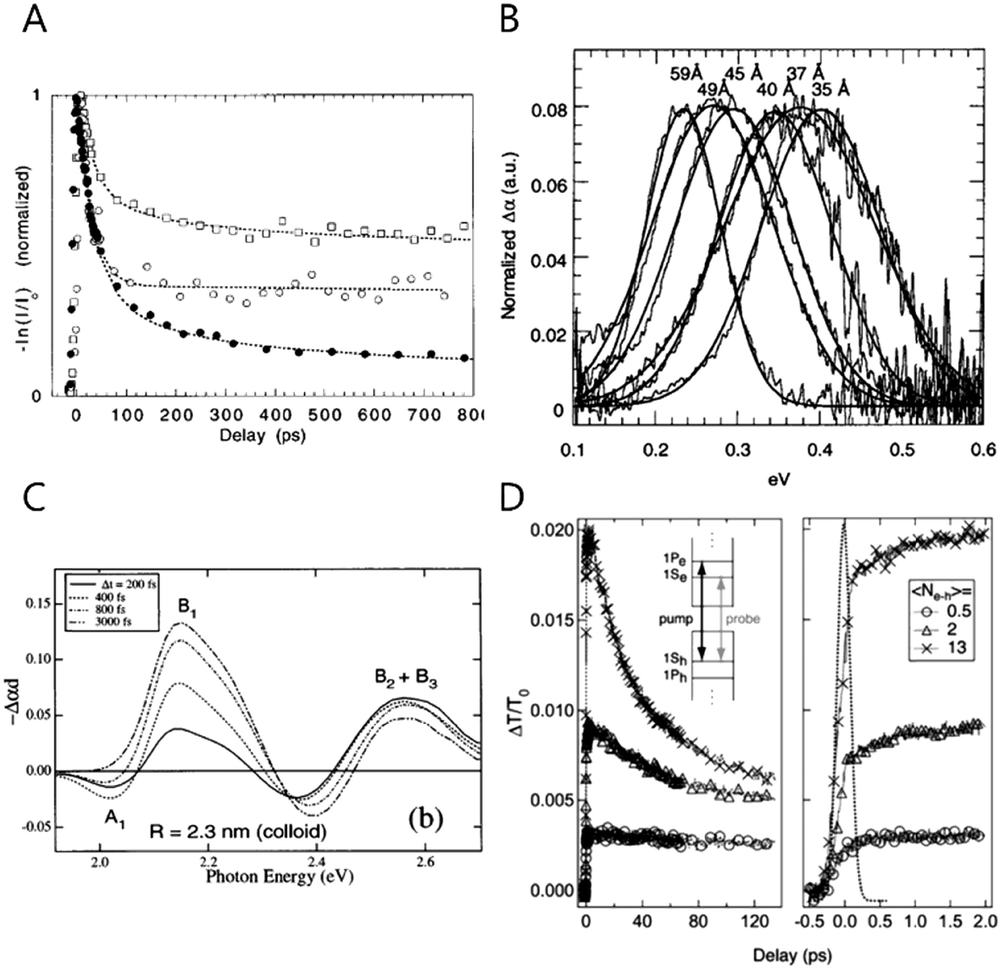 | ||
| Fig. 1 (A) The CdSe NC time evolution of the visible-induced IR absorption spectra with size dependence filled circles 43 Å, open squares 38 Å, open circles 31.5 Å respectively. Reprinted with permission from ref. 22 Copyright (1998) by the Applied Physics Letters, (B) room temperature visible induced transient FTIR spectra reprinted with permission from ref. 17. Copyright (2000) by the American Chemical Society, (C) TA spectrum of different delay between delay and pump time with different size of NC. Reprinted with permission from ref. 25. Copyright (1998) by the American Physical Society, (D) room temperature population dynamics of 1S state with photo-excited carriers. Reprinted with permission from ref. 18. Copyright (2005) by the American Physical Society. | ||
To make the intraband transition useful for applications, the lifetime should be slower than the pico- or femtosecond time scale. Hence, there have been many efforts to achieve long-lived carriers in the CB such as the bandgap engineering of the core/shell nanocrystals.11,12,24,27,28 Elongating the carrier lifetime has been successful to some degree by several researchers studying with bandgap-engineered nanocrystals, but the time scale is still too fast to use for applications27 Furthermore, the intraband radiative recombination process was not possible to observe in those days.
Electrochemical doping
In order to observe the intraband transition without using the ultrafast spectroscopy, electrochemical potential or chemical treatment method were proposed by the Guyot-Sionnest and his co-workers in 2000 and 2001, providing a method to observe the intraband transition using a commercial infrared spectrometer in steady state, which also renders opportunities to perform the trion or biexciton studies.17,19In electrochemical doping, the quantized electronic states, mainly the 1Se state of the nanocrystal, are filled with carriers injected through the electrochemical method in the electrolyte.19,29 The nanocrystals in the film are crosslinked with each other through sufficiently short ligands to allow conductivity, and an external potential is applied to the nanocrystal film through electrochemical gating effect. The applied potential varies the Fermi level of the nanocrystal, thereby manipulating the density of carriers inside the nanocrystal. Raising (or lowering) the Fermi level with the electrochemical potential allows stable occupation of electrons (or holes) in the electronic state, so that the electronic states of the quantum dots beyond the 1Se(1Sh) state can be readily explored. The higher electronic states are monitored by optical absorption or conductivity change as shown in Fig. 2D and E.30 Compared to the field effect transistor, the electrochemical doping method has an advantage of charging the entire quantum dot film since the electrolyte efficiently penetrates into the quantum dot film whereas the field effect transistor charges only a few monolayers nearby the dielectric layer.31,32 The electrochemical doping method has been rapidly developed and enabled sophisticated control of the luminescence of nanocrystals. For example, the variation of the potential leads to the intensity change of the bi-color emission from the nanocrystal and the matrix mixture.33 Furthermore, studies that employed the electrochemical doping method for nanocrystals provided valuable information on the dynamics of carriers and higher quantum states of nanocrystals, leading to the fundamental understanding of nanocrystals.29,34
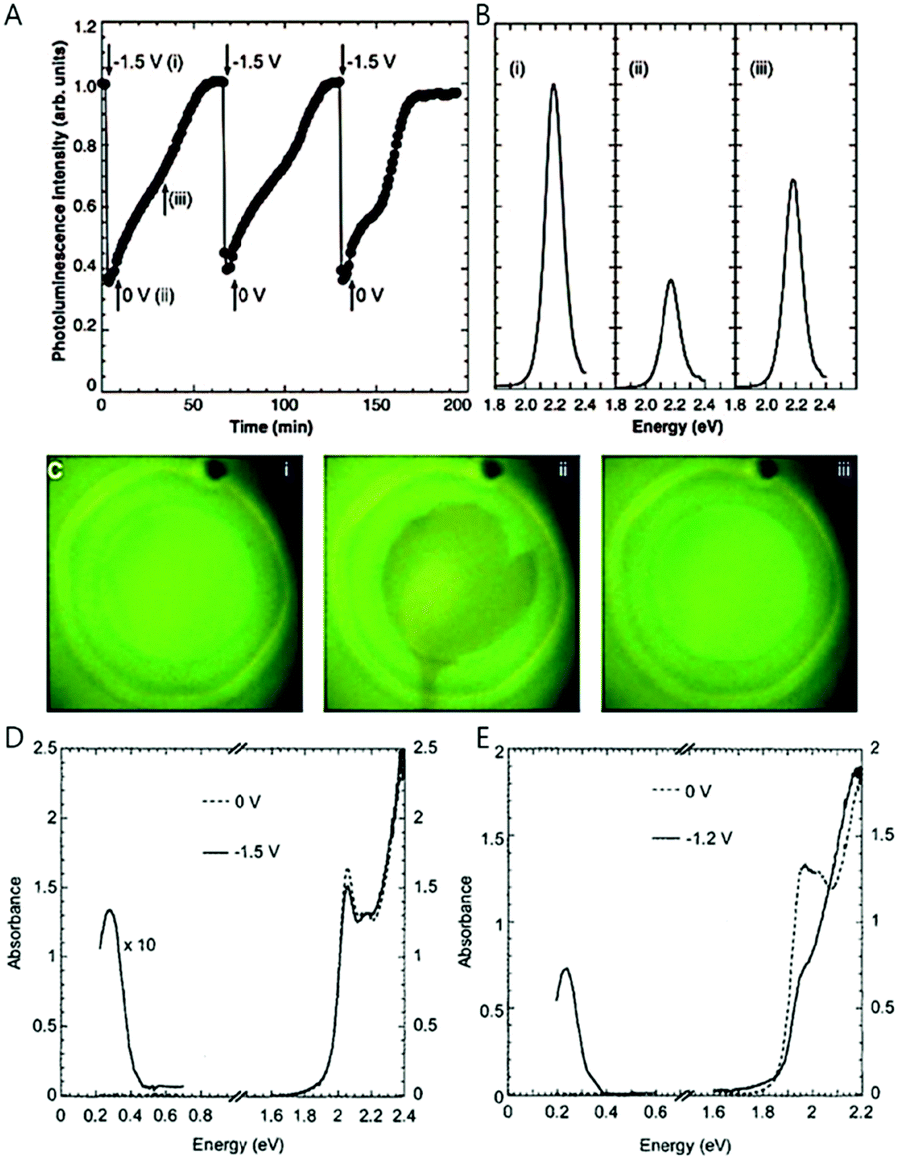 | ||
| Fig. 2 (A) Integrated photoluminescence intensity of CdSe/ZnS with different potentials. (B) Photoluminescence spectra with different potential (C) sample photograph with UV illumination; 0 V, −1.5 V, and resetting to 0 V respectively. IR and UV-visible spectra of (D) 5.4 nm CdSe nanocrystals and (E) 7.0 nm CdSe nanocrystals with different potentials from ref. 19. Reprinted with permission from AAAS. | ||
Although electrochemical doping is an excellent method for the fine control of doping density in nanocrystals, it does not ensure permanent high doping density of the nanocrystal since the nanocrystal is susceptible to decomposition by the applied potential in some cases. The stable potential range is an indicator that shows the electrochemical potential range in which the material can stably maintain the excess carriers. The stable potential range can be determined by considering the reduction potential of the constituent elements of the nanocrystals. For example, the range of stable doping for HgS lies between −0.73 V and 1 V, which is obtained from the oxidative and reductive reaction of HgS bulk according to ref. 35. The oxidative potential for (HgS)n + 2h+ → (HgS)n−1S + Hg2+ is 1 V/NHE using E = ΔG0f/2F + E0, where ΔG0f, F and E0 are the free energy of formation, Faraday constant and reduction potential, respectively. Therefore, if the holes are generated at a potential more positive than 1 V, the HgS nanocrystals is readily decomposed (oxidized). Similarly, when the electrons are present at a potential more negative than −0.73 V, the HgS nanocrystals are easily reduced. For ZnSe nanocrystals with a reduction potential of −0.762 V (vs. SHE) for Zn/Zn2+, it is less likely to form an n-doped material because the conduction band of the bulk ZnSe is −1.5 V (vs. SHE), meaning that the material can be readily oxidized by giving off excess electrons. Furthermore, the electrochemical doping is likely to have a tolerable number of oxidative/reductive charging cycles due to the possibility of the surface chemistry change.
Charge transfer doping
Charge transfer doping, which has been frequently used in the field involving conducting polymers,36 was first successfully applied to colloidal quantum dots in 2000.17 The addition of a reducing agent such as sodium biphenyl causes the injection of electrons to nanocrystals, resulting in the occupation of the lowest electronic state (1Se) of the CB with electrons under inert conditions. As the 1Se state is fully filled, the intraband transitions (1Se–1Pe) of the electrons and the corresponding bleach of interband transition (1Sh–1Se bandgap) appear in the mid infrared and visible region, respectively (Fig. 3A and B). This method has been used for understanding the quantized state of nanocrystals as shown in Fig. 3B.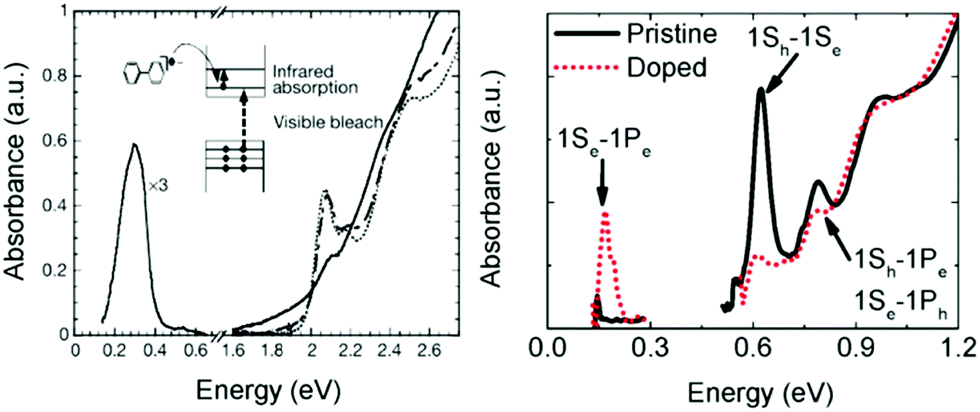 | ||
| Fig. 3 (A) Absorption spectra of CdSe NC (dotted line) with biphenyl reagent (solid) and without (dashed line) from ref. 17. Reprinted with permission from AAAS. (B) Pristine and cobaltocene treated PbSe absorption spectra from ref. 37. Reprinted with permission from AAAS. | ||
Unfortunately, the charge transfer doping method is restricted to inert conditions since the carriers are readily quenched upon air exposure (Fig. 3A). Therefore, the charge transfer doping heavily relies on the reduction potential of the reducing agent and the size of the nanocrystal. Nonetheless, the charge transfer doping approach has been frequently used to dope the nanocrystals or to explore higher electronic state.37
Other doping methods such as photoelectrochemical doping are also available, but those methods were already fully covered by other excellent review papers.38 Thus, we focus on the doping methods that were incorporated for studies involving the intraband transition.
Self-doping: mercury chalcogenide nanocrystals
The methods described above have several limitations when used for studies on the intraband transition. For example, the ultrafast spectroscopy enables one to observe the intraband transition only in the fast time scale. Also, the intraband transitions are only observed when either an electrochemical potential is applied or reducing agents are present under inert condition. Indeed, it has been difficult to dope nanocrystals without a post-doping process since the carriers swiftly go through recombination processes once added. Additionally, the carriers are prone to involve in chemical reactions occurring at the surface of the nanocrystals.The synthesis of the self-doped (or lightly doped) nanocrystals where a high doping density is achieved without the use of any external factor such as an electron-donating molecule, applied electric field, or optical pumping, has great significance for the nanomaterial research utilizing quantum confined states. In colloidal quantum dot applications, only the bandgap transition has been intensively utilized for applications, although there are a number of possible transitions occurring over the inter- and intraband. The self-doping approach will offer more opportunities to fully exploit the quantum confined states (Fig. 4A and B).
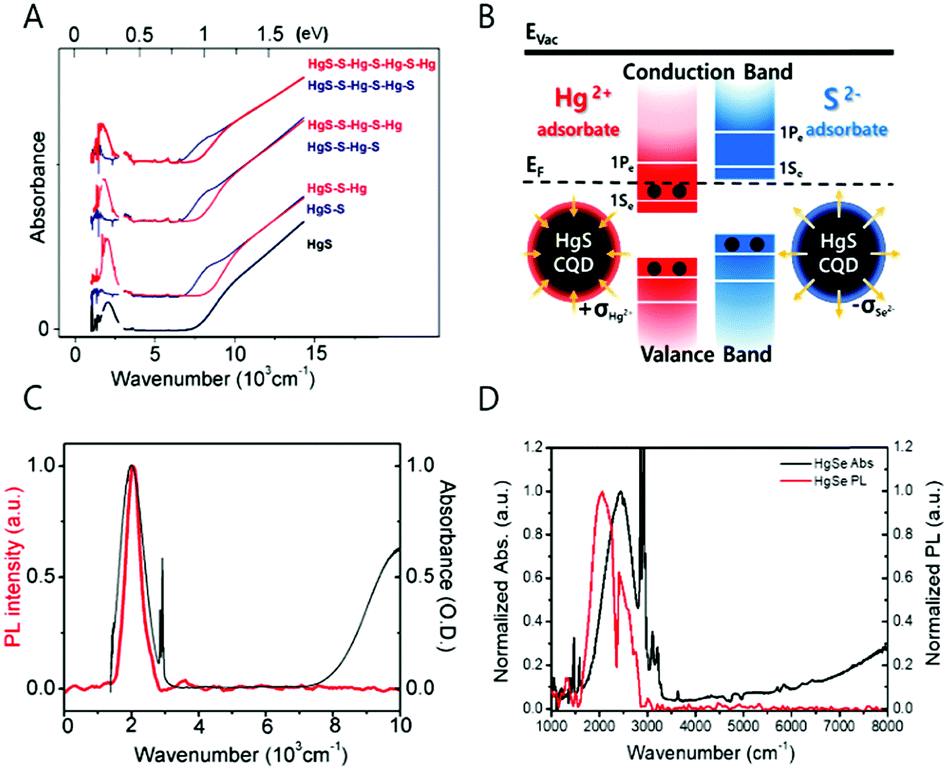 | ||
| Fig. 4 (A) Absorption spectra of HgS CQD with S2− and Hg2+ layer by layer growth. Reprinted with permission from ref. 36 copyright 2014 American Chemical Society. (B) Surface treatment schematics of HgS CQD (C) HgS absorption and photoluminescence spectra reprinted with permission from ref. 39. Copyright 2014 American Chemical Society. (D) HgSe absorption and photoluminescence. | ||
Recent progress in the studies of the intraband transitions by our own group and the Guyot-Sionnest group made observation of the mid-IR intraband transition in steady state possible by successfully incorporating excess electrons into the CB, providing a way to exploit the mid-IR electronic transition for fundamental understanding of the physical property of hot carriers and for various optoelectronic applications.39 The steady state intraband transition has been realized due to the electron filling of the lowest electronic state (1Se) of the CB (Fig. 4B and 6A). By controlling the number of electrons in the nanocrystal through chemical synthesis without an addition of heterogeneous metal impurity, it is possible to tailor the magnetic and optical property of the nanocrystal.39
Understanding the mechanism of how electrons fill the electronic states of the CB is useful for many reasons. Firstly, it provides clear understanding of the electron dynamics in the nanocrystal by excluding the hole dynamics occurring in the VB or in the mid-gap states. In addition, it also allows the intraband transition in steady state without worrying about the bandgap-based Auger process.
Secondly, the ability to control the number of electrons in the discrete energy states offers a way to tailor the magnetic properties determined by the number of unpaired electrons. The magnetic property depending on the number of unpaired electrons in the nanocrystal is different from that resulting from the paramagnetic property of the transition metal-doped semiconductor nanocrystals.40,41 The magnetism study of the self-doped nanocrystal will provide useful insight to understanding the nature of the spin of the carrier in nanocrystals. Furthermore, spintronics and quantum computing applications can be considered for future applications using nanocrystals with unpaired electrons.
To note, the self-doped nanocrystals can serve as promising materials along with other materials generating long wavelength light emission. Graphene and metal nanomaterials researchers have carried out intensive studies to produce emission through various methods.42–47 For example, graphene nanoribbons create a narrow bandgap and show emission in the infrared.42–44,48 In addition, the metal clusters are interesting due to the unexpected emission in the visible regime. Generally, nanomaterials with a high density of charge carriers exhibit surface plasmon resonance (SPR) and thermal emission under photoexcitation. The fact that the excitonic radiative recombination of these materials were observed indicates the emergence of holes by tuning the size or shape.49,50 In other words, the concentration of charge carriers was intentionally lowered via modifications, so that excitonic radiative recombination was possible. With respect to changing the level of carrier density, the intraband transition study uses an approach that is opposite to that used by studies involving graphene and metal nanomaterials. At a low level of carrier density similar to that of a typical semiconductor nanocrystal, the intraband transition of the semiconductor nanocrystals is very fast (picosecond time scale) followed by the bandgap transition. Thus, colloidal quantum dots of relatively low carrier density require additional carriers to be able to stably fill the lowest electronic states of the CB.
Neither the optical property nor the magnetic property of the self-doped nanocrystals was successfully observed before 2014. To note, the doping density achieved by the self-doping approach to be discussed is smaller than that of the heavy doping regime, so there are other empty quantized states remaining in the CB. The electron density of the self-doped nanocrystal is less than 8 per nanocrystal.24,39,40,51 The appearance of the intraband transition measured in steady state is clear evidence of the successful self-doping because the steady state 1Se–1Pe intraband transition would not have been observed if the lowest electronic states of the CB, 1Se, are empty of electrons (Fig. 4B). The first report on semiconductor nanocrystals self-doped with electrons was shown with mercury sulfide colloidal quantum dots by Jeong et al. in Fig. 4C.39 The black and red spectra correspond to the absorption and emission spectra of the steady state intraband transitions of HgS CQDs, respectively and the wavelength of the spectra relies on the nanocrystal size. The dip of the emission spectrum and the sharp peaks of the absorption spectrum at 2890 and 2910 cm−1 are attributed to the symmetric and asymmetric CH vibrational modes of the dodecanethiol ligands. The infrared photoluminescence is quenched by the vibrational mode of organic molecules. Regarding the synthesis process, there are no challenging synthetic techniques involved to generate the self-doped mercury chalcogenide colloidal quantum dot. The HgS and HgSe colloidal quantum dots are synthesized with no difficulty by modifying the procedure for synthesizing HgTe colloidal quantum dots.52,53 However, it is worth to note that one should handle the mercury compound with great care during the synthesis due to the extreme toxicity.
Since the intraband transition is available in steady state under ambient condition, the electronic transition can be used for photodetection through exciton splitting. The intraband photocurrent of the HgE quantum dot film has been detected in the mid-IR regime.31 To note, the intraband transition is explicitly different from the localized surface plasma resonances (LSPRs) in that the intraband transition produces radiative recombination under photoexcitation whereas the LSPRs undergo no excitonic emission. More information on the work involving the use of intraband transition for mid-IR photodetection can be obtained by excellent reviews written by Guyot-Sionnest and Lhuillier.54
Interestingly, the self-doped colloidal quantum dot is highly sensitive to the charge of the nanocrystal surface as shown in Fig. 4B. Identical constituent ions of the nanocrystals were added into the HgS colloidal quantum dots.39 Surprisingly, the addition of the anion (S2−) and cation (Hg2+) leads to quenching and recovery of the intraband transition, respectively. Specifically, the ammonium sulfide in formamide solution was added to the HgS CQD in tetrachloroethylene solution, which led to the quenching of the intraband absorption and photoluminescence. In contrast, consecutive addition of the mercury cation recovers the intraband transitions. The intensity variation of the intraband transition is reproducible under ambient conditions. The appearance and disappearance of the intraband transition is attributed to the surface dipole of the nanocrystal.39 Concretely, the positive charge at the surface balanced by the surrounding counter anion lowers the electronic states of the nanocrystal versus vacuum level, eventually leading to the appearance of the steady-state intraband transition. In contrast, the negative charge on the surface balanced by the surrounding cation results in the elevation of the electronic state, resulting in the disappearance of the intraband transition and recovery of the bandgap transition.
The electrochemical result supporting this hypothesis is well described in the ref. 39. Both conduction and valence band energy states of HgS, HgSe and HgTe nanocrystals are largely perturbed by the post surface treatments. Both homogeneous and heterogenous ions were used to generalize the effect of the cation and anion surface potentials.
In the microscopic view, the shift of the conduction and valence bands can be explained by the change of the surface dipole on the nanocrystal surface.24 In the sulfide treated HgE CQDs, the sulfide (S2−) binds to the mercury ions at the surface of the nanocrystals. Therefore, the surface dipole pointing outward from the bound sulfur elements to free positive ions surrounding the nanocrystal causes both conduction and valence band energies to go up, making HgS CQD undoped. Meanwhile, the surface dipole of the mercury ion treated HgS CQD points inward from the free negative ions to the bound mercury elements at the nanocrystals, leading to the n-doped nanocrystal through lowering the band energies (Fig. 4B). The electrostatic potential at the surface area of 20 Å2 of (111) was roughly calculated to be 1.8 eV with the smallest dielectric screening being ε = 1. The potential change caused by the surface dipole is very large compared to the bandgap of the nanocrystals at ∼1.0 eV. This hypothesis is supported by the surface sensitive work function of the metal induced by self-assembled monolayers.55
The variation of the conduction and valence band energies has been observed by others as well. Brown et al. measured the shift of the band energy of PbS colloidal quantum dot with various surface ligands by using ultraviolet photoelectron spectroscopy. It was confirmed that the band energy is readily altered by varying the surface conditions.56 The surface dipole dependence of the band energy may also explain the enhanced device performance of the quantum dot film treated with cations. Wang et al. presented the variance of the conductivity of PbS quantum dot film during successive ionic layer adsorption and reaction (SILAR).57 The enhancement in the performances probably originate from the lowering of both the conduction and valence band energy, leading to a large doping density of the n-type quantum dot.
With respect to the material stoichiometry, the stoichiometry of the nanocrystals is apparently changed by the addition of the anion and cation resources. Based on the result of inductively coupled plasma optical emission spectroscopy (ICP-OES) and the XPS analysis, the as-synthesized or the mercury ion treated HgS CQDs show ∼30% excess of the cation while the sulfide treated HgS CQD has an equivalent amount of the metal and chalcogenide.23 The excess metal proportion is frequently observed from other n-type colloidal quantum dots such as PbS and PbSe.5,37,58 However, these nanocrystals do not generate steady state intraband transitions because the 1Se state lies higher than the H2/H2O reduction potential, resulting in the loss of the electron of the 1Se by the hydrogen gas generation.59,60 To note, the stoichiometrically balanced core/shell quantum dots such as HgSe/ZnS CQD do not appear the steady state intraband absorption, that could limit the use of the transition for further applications.
The observed steady state intraband transition of the colloidal quantum dot has mostly been viewed as the 1Se–1Pe transition.23 Due to the fast relaxation of the electron, we barely achieved to observe the first intraband transition (1Se–1Pe) of CB in steady state. If further carriers were to occupy the 1Pe state in steady state, the second intraband transition (1Pe–1De) should appear in principle.
Conventionally, thiol ligand molecules have been incorporated during the HgE (E = S, Se) nanocrystal synthesis. However, in order to get rid of any possible concerns including hole quenching or oxidation process under IR photoexcitation, we used oleylamine ligands rather than the thiol ligands (Fig. 5A and B). Surprisingly, the oleylamine passivated HgS CQD films on a gold substrate demonstrate both the first and the second intraband transitions (Fig. 5C and E). Although there was no success of removing the first intraband transition (1Se–1Pe) by complete carrier occupation of the 1Pe state, the emergence of the second intraband transition offers a new avenue for future optical property study of colloidal quantum dots. The longitudinal optical (LO) phonon of the HgS nanocrystal was measured by using Raman spectroscopy, and the value of LO phonon is 262 cm−1 (32 meV), which is one order of magnitude smaller than the intraband transition energy. The second lowest intraband transition with the amine-based ligands will be promising for the use of the nanocrystal for multistep memory and flexible device (Fig. 5C–F).
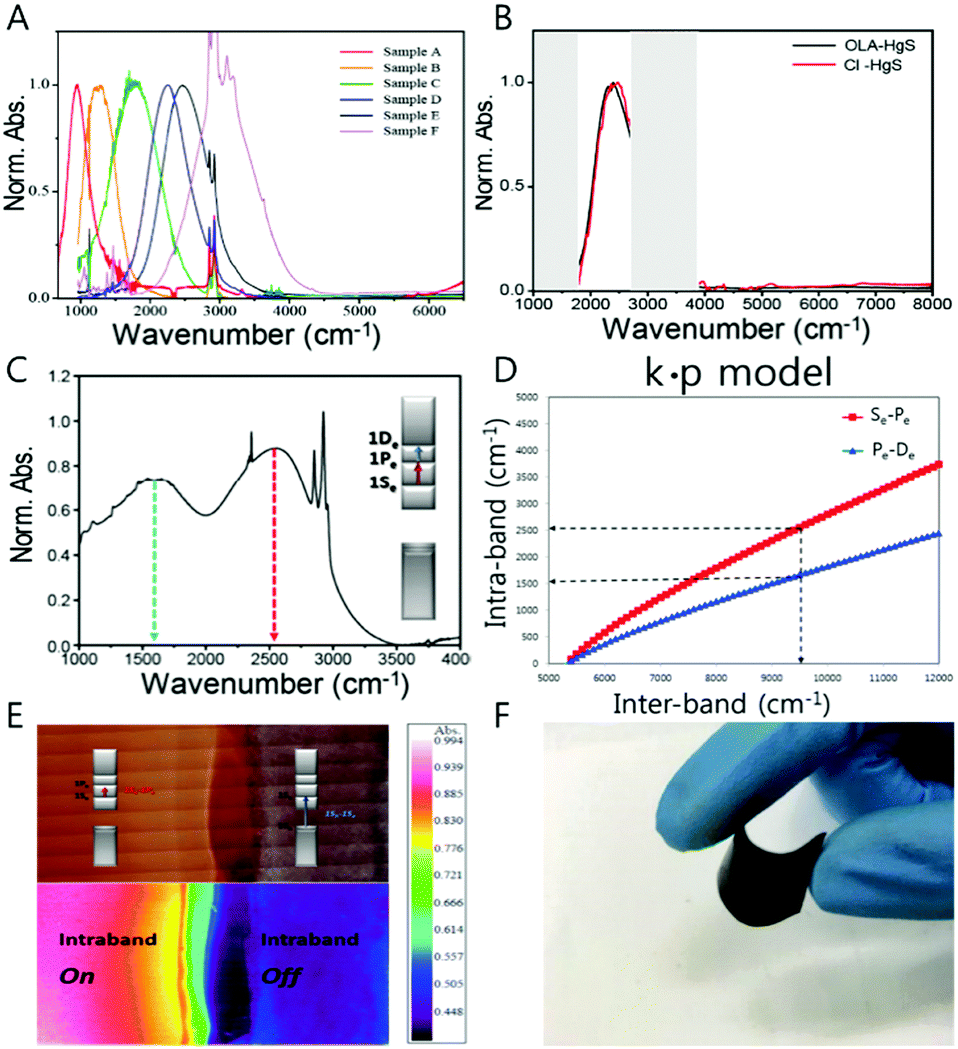 | ||
| Fig. 5 (A) Absorption spectra of HgS CQD (B) HgS absorption with oleylamine and atomic ligand (C) 1Se–1Pe and 1Pe–1De absorption spectrum (D) intraband and bandgap correlation-based k·p model (E) IR microscope image of doping controlled HgS permission from ref. 24. Copyright 2016 American Chemical Society. (F) Flexible HgSe CQD film. | ||
The mercury chalcogenide colloidal quantum dot showing the intraband transition under steady state was thought to be occupied by two electrons in the 1Se state. As a result, no distinct magnetic feature was observed from the HgS CQDs due to the paired electrons unless metal impurities such as manganese are incorporated into the nanocrystals20,61,62 If so, before fully occupying the 1Se state, an intermediate state with single electron occupation should be present, which is conceptually analogous to the singly occupied molecular orbital (SOMO). Our results proved the existence of the singly occupied quantum state (SOQS), achieved by kinetically controlling the chemical synthesis process.40 (Fig. 6A, D and E)
 | ||
| Fig. 6 (A) Schematic diagram of electron occupation of conduction band (B) XPS and (C) XRD data of HgSe (D) EPR signal and (E) absorption spectra of HgSe (F) the EPR signal of HgSe before (red), after (blue) and as synthesized (black). Adapted with permission from ref. 40. Copyright 2017 American Chemical Society. | ||
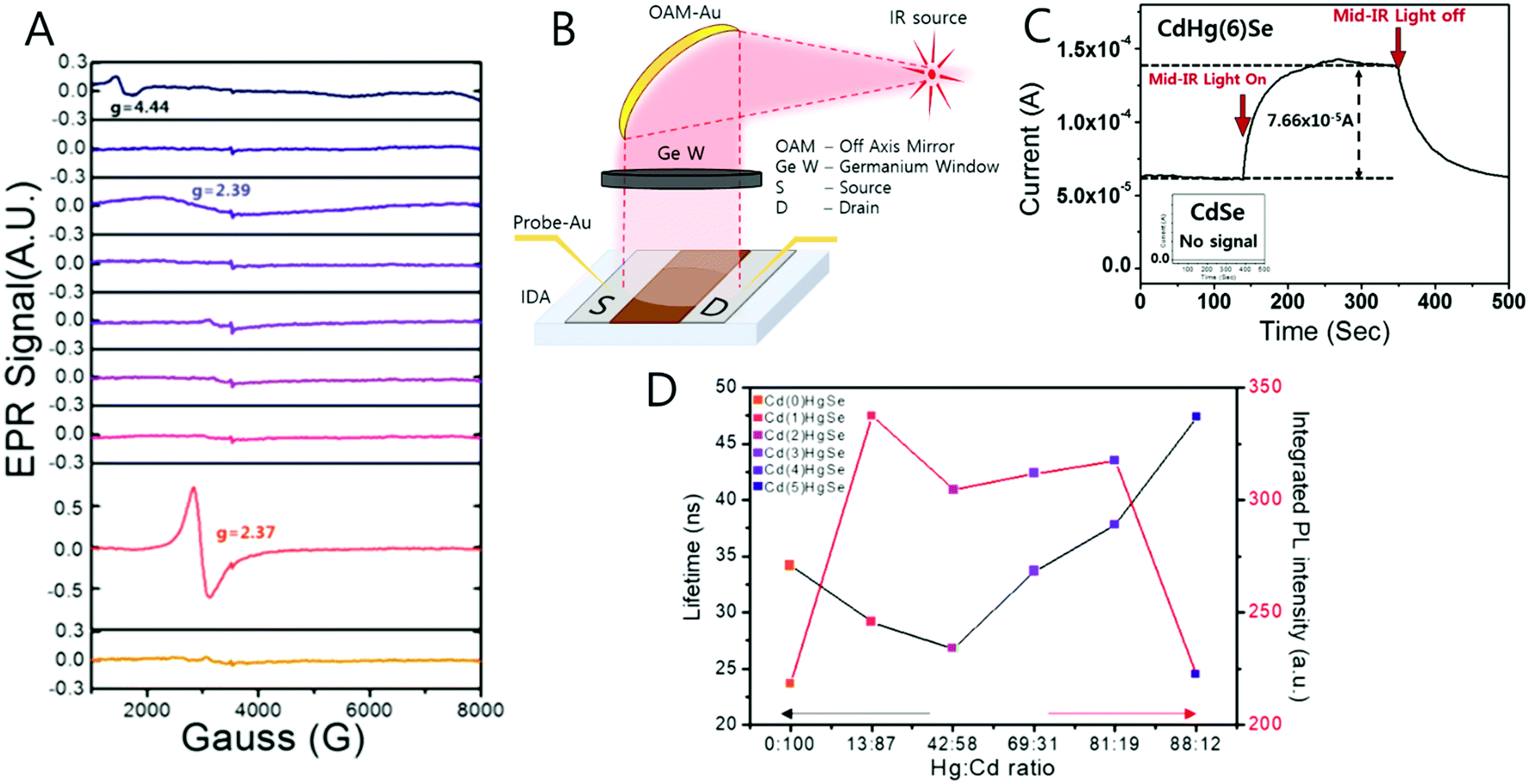
The unpaired electron at the 1Se state is detected by electron paramagnetic resonance (EPR) spectroscopy that has a g-value of 2.54, which is larger than that of free electrons. In semiconductors, the g-value of the carrier relies on the carrier density (Fig. 6D and F). Therefore, depending on the electron density tuned by changing the size of the nanocrystal, the g-value can slightly change. The stoichiometry of the nanocrystal largely affects the emergence of the SOQS. For example, the SOQS arises only when the metal-to-chalcogenide ratio is over unity and when smaller than 0.3. When the metal-to-chalcogenide ratio is around 0.3, the 1Se state seems to be fully filled with two electrons referred to as the doubly occupied quantum state (DOQS) (Fig. 6A–C). Surprisingly, the HgSe nanocrystal in the solid state ensemble showing the paramagnetic property produces different g-values at different angles to the magnetic field. The peak corresponding to the unpaired electron shifts to lower Gauss at both 90° and 270°, and recovers its original Gauss at 180° (Fig. 7B). This infers that the spins of the nanocrystals have preferred direction when the samples are dried under vacuum.
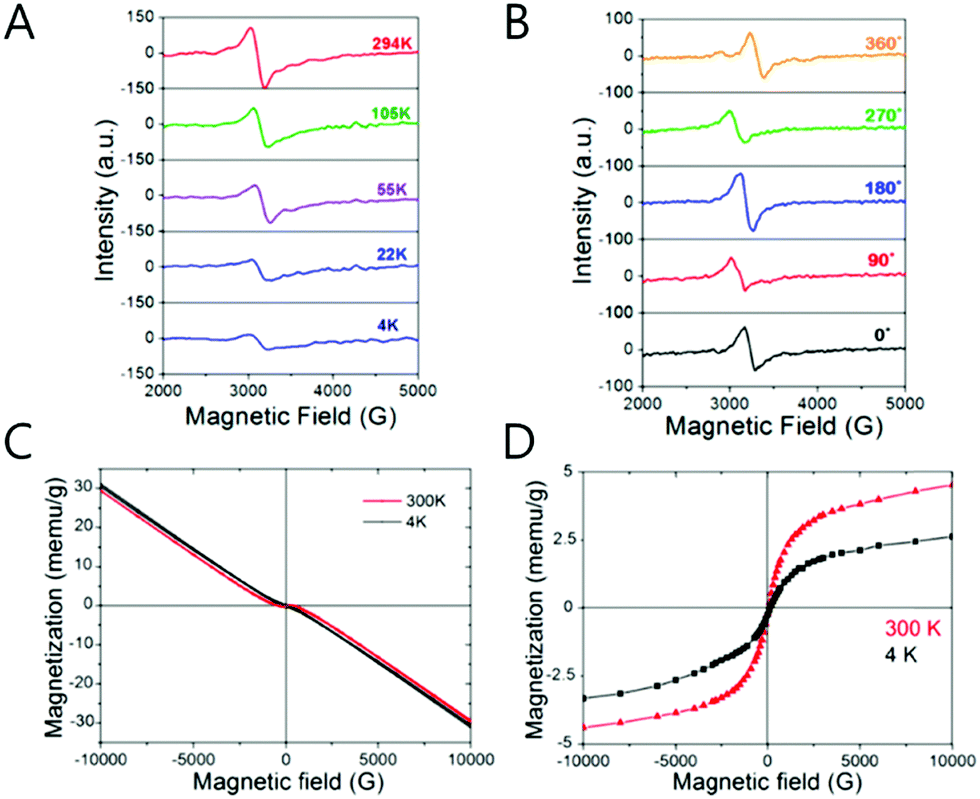 | ||
| Fig. 7 (A) The temperature dependent EPR spectra of SOQS HgSe (B) angle dependent EPR spectra of SOQS HgSe (C) magnetization curves of HgSe CQD with different temperature (D) superparamagnetic components with 300 and 4 K temperature. Adapted with permission from ref. 40. Copyright 2017 American Chemical Society. | ||
The XRD spectra indicate that the HgSe(111) facet is enhanced when the nanocrystal exhibits the steady state intraband transition (Fig. 6C). This carefully suggests that a geometric distortion arising from the enhancement of a certain facet may be required for the steady state intraband transition.
The magnetization of the film of the HgSe nanocrystal has been investigated by the superconducting quantum interference device (SQUID) measurement (Fig. 7). The HgSe films demonstrate the superparamagnetic and diamagnetic feature for the SOQS (Ne = 1) and DOQS (Ne = 0 or 2), respectively40 (Fig. 6D). To note, no heterogeneous metal impurity was added into the nanocrystal. Thus, the magnetic property examined are intrinsic of the HgSe nanocrystals. Due to the large surface-to-volume ratio, the electronic property swiftly responds to the surface environment. Just as the intensity of the intraband transition is tuned by the surface treatment, proved in previous papers, the magnetic property is also governed by the surface treatment. (Fig. 6F) Further magnetic properties of the HgSe CQDs arising from the occupation of the higher electronic state were also reported, although more thorough study is required to assign the specific electronic state where the unpaired electron remains.
The electron occupation of the electronic state of the CB also seems to enhance the electron mobility. An electron mobility of 1.29 cm2 V−1 s−1 was obtained from the Al/SCN-HgS CQD/SiO2/Si field-effect-transistor (FET). The intraband transition of the HgS CQD film attenuated through the annealing process due to the efficient coupling of nanocrystals. The optimum annealing temperature of 90 °C for the HgS CQDs was found as well, which is reasonable considering the nanocrystal reaction temperature of 120 °C. Although the self-doping carrier density is larger than the conventional quantum dot, the electron mobility was not substantially enhanced.64
Along with the nanocrystal size, the compositional change of the nanocrystal significantly affects the electronic transition. The cation-exchange method where the portions of Hg and Cd are varied in the CdxHg1−xSe alloy nanocrystal was introduced to tune the major electronic transition from the visible to mid-IR of nanocrystals.63 (Fig. 8A) Through the compositional change, we succeeded in shifting the major electronic transition from the bandgap transition to the intraband transition. In addition, the localized surface plasmon resonance (LSPR) was observed as well due to the increase in the carrier density of the nanocrystal (Fig. 8B and C).65–67 The change of the major electronic transition from the bandgap transition to LSPRs represents that the nanocrystal properties dramatically change from the semiconductor to metal-like nanocrystal through the cation exchange method.
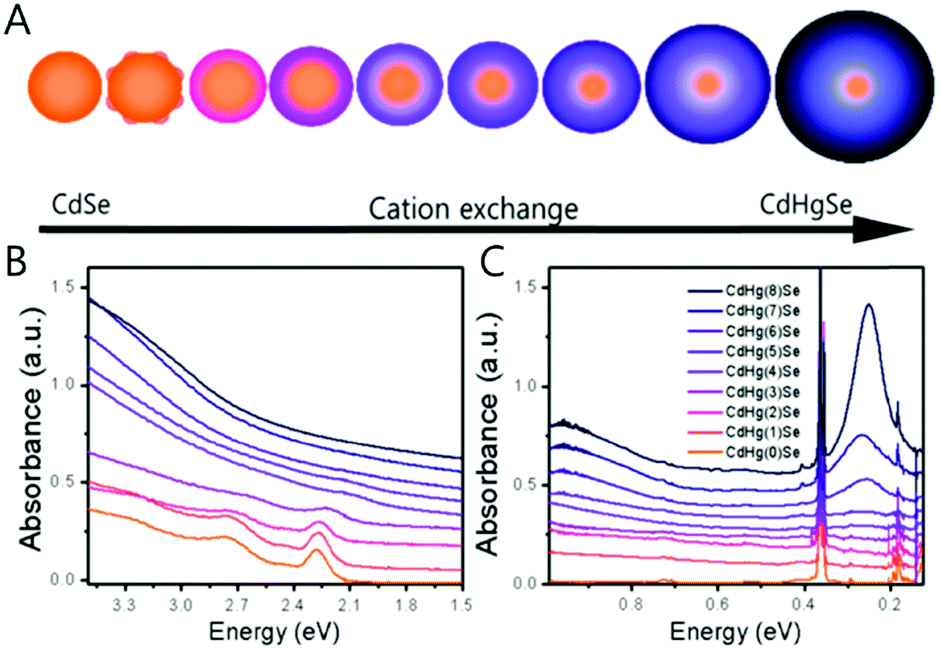 | ||
| Fig. 8 (A) Schematics of CdSe to CdHgSe cation exchange (B) UV visible and (C) FTIR absorption spectra of CdHgSe cation exchange series. Adapted with permission from ref. 63. Copyright 2017 American Chemical Society. | ||
During the compositional change, the corresponding magnetic properties were carefully monitored by EPR spectroscopy in ambient conditions (Fig. 9A). Interestingly, the magnetic property varies from diamagnetic to paramagnetic and again recovers the diamagnetic property. The appearance and disappearance of the paramagnetism depending on the portion of Hg in CdxHg1−xSe alloy nanocrystal reveal that the lifetime of the unpaired carrier is not negligible even for the alloy nanocrystal formation process (Fig. 9D). The electron dispersive X-ray (EDS) elemental mapping (Fig. 10) clearly visualized the compositional change of the CdxHg1−xSe alloy nanocrystal. As the major electronic transition is tuned from the bandgap to the intraband transition, the nanocrystals respond to different wavelength irradiation. For example, with increased Hg element of the alloy nanocrystal, the major electronic transition appears in the mid-IR regime, and thereby the mid-IR irradiation generates the corresponding photocurrent. On the other hand, the CdSe nanocrystal, the starting material, exhibits no photocurrent upon the mid-IR irradiation (Fig. 9B and C). When the carrier density exceeds the doping range leading to a substantial amount of electrons in the CB, the LSPR can appear in the mid-IR regime as reported by Shen et al.68 The same feature was identified in the CdxHg1−xSe alloy nanocrystal. Manipulating the composition of the alloy nanocrystal is a promising method to control the optical, electrical and magnetic properties caused by tuning the carrier density in the nanocrystal. The cation exchange and successive nanocrystal growth allow the expansion of the range of electronic transition from visible to mid-IR range in this work. In principle, nanocrystals with shorter bandgap transition energy such as blue visible light or up to ultraviolet should also be possible to be tuned down to the terahertz (THz) range with optimum combination. With respect to applications, the absorption spectra of the CdxHg1−xSe alloy nanocrystal has optical selectivity compared to the bulk commercial infrared detectors with a broad absorption spectrum such as CdHgTe (MCT).
 | ||
| Fig. 9 (A) EPR spectra of CdHgSe series measured in room temperature (B) the schematic photocurrent measurement setup and (C) photocurrent response and (D) lifetime and photoluminescence relation. Adapted with permission from ref. 63. Copyright 2017 American Chemical Society. | ||
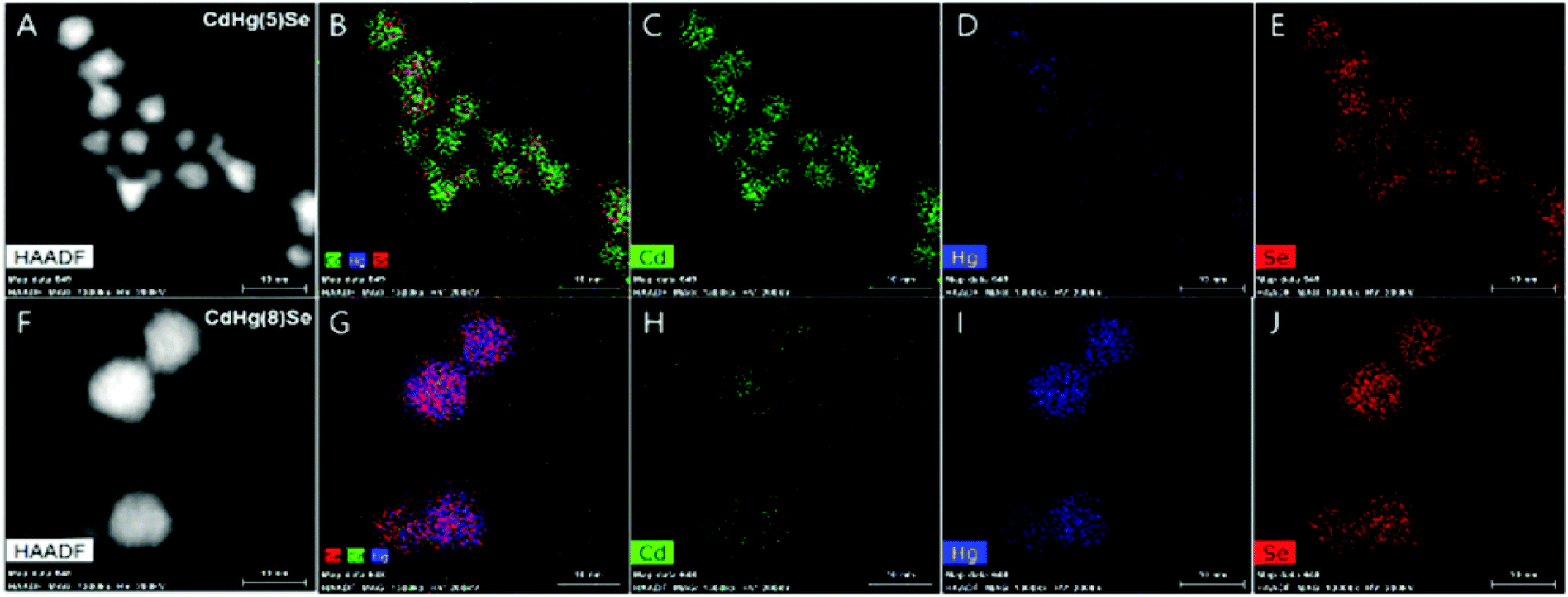 | ||
| Fig. 10 High-angle annual dark-field (HAADF)-STEM image of CdHg(5)Se (A–E) and CdHg(8)Se (F–J) CQD Cd (green), Hg (blue), and Se (red). Adapted with permission from ref. 63. Copyright 2017 American Chemical Society. | ||
Silver chalcogenide: toward non-toxic self-doped nanocrystals
The mercury element in the nanocrystal exhibiting the intraband transition in steady state has been indeed a barrier for researchers who have little experience with handling the mercury compound. Therefore, there is a great need to replace the Hg with a less toxic or non-toxic material. We, therefore, synthesized the Ag2Se colloidal quantum dot that produces a mid-IR absorption spectrum.69 The synthetic method of the Ag2Se is not significantly different from what Sahu et al. reported.70,71 Sahu et al. reported that the confinement is weak based on the small shift of the mid-IR absorption peak when varying the nanocrystal size, which they attribute to the high carrier density of the Ag2Se nanocrystal.71 To note, the observed mid-IR absorption peak was assigned as the bandgap transition by Sahu et al. Our spectroelectrochemistry results demonstrate that the mid-IR absorption peak emanates from the intraband transition while the corresponding bandgap transition appears at the NIR regime (Fig. 11C and D). Both transitions are strongly correlated with each other, corroborated by the fact that when one transition is enhanced, the other is attenuated since the 1Se state is shared during the electron transition. It is important to note that the electron occupation of the 1Se state is only observed when the metal element is in excess in the Ag2Se nanocrystal. Under photoexcitation, the intraband exciton is created in the electronic state of the CB, and the mid-IR radiation is emitted, which is a direct evidence for the formation of the intraband exciton (Fig. 4D and 11A). The intraband PL is also size-tunable as similar to the bandgap PL since the electronic states in the CB are affected by the quantum confinement effect as well. The exciton formation of the Ag2Se CQD is confirmed by the mid-IR photocurrent measurement. The Ag2Se/ZnO thin-film transistor generates a photocurrent under mid-IR irradiation and the threshold voltage shifts to negative values, confirming the exciton splitting of charge carriers.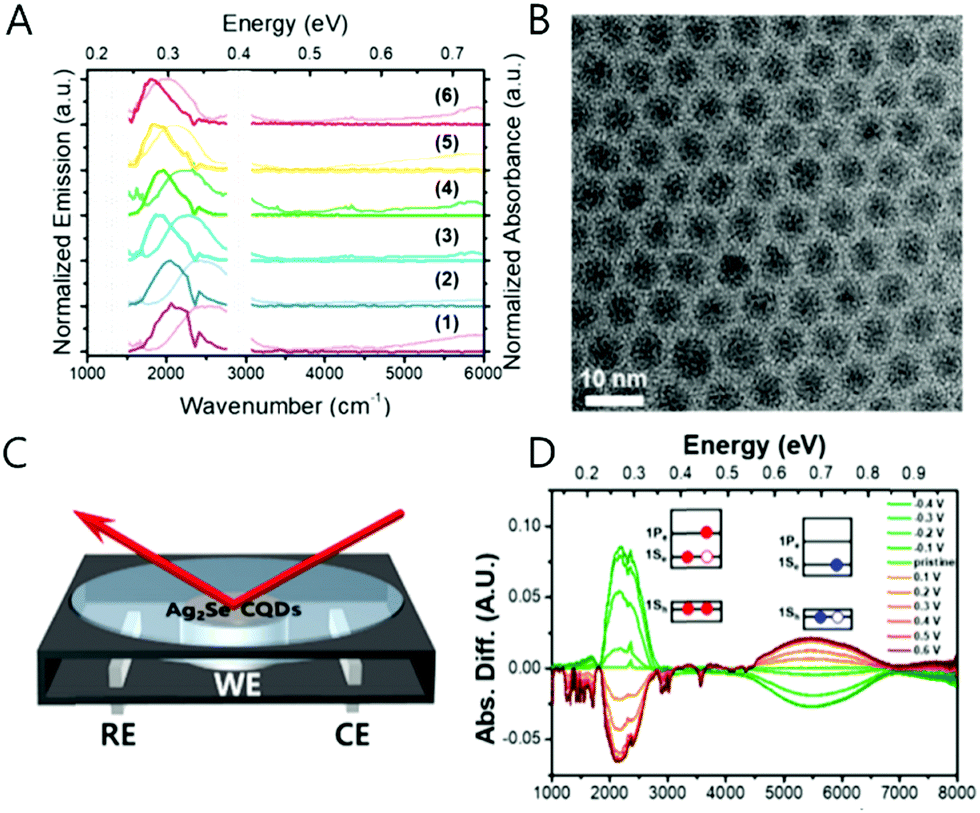 | ||
| Fig. 11 Ag2Se CQD absorption and photoluminescence (A) and TEM image (B). The schematics of spectro-electro chemical cell (C) and reduction (green) and oxidation (red) spectra of Ag2Se sample (D). Adapted with permission from ref. 69. Copyright 2018 American Chemical Society. | ||
General methods to generate the intraband photoluminescence
It has been only four years since the first observation of the intraband PL of the colloidal nanocrystals. In 2014, 2016, and 2018, the method to observe the intraband PL of HgS, HgSe, CdSe and Ag2Se CQD were reported by the Guyot-Sionnest and Jeong groups.10,22–28,31,32,34,39,40,63,69The intraband PL for cadmium chalcogenide colloidal quantum dots was measured in 2016, inferring that the intraband PL can be a general feature of the colloidal nanocrystals.27 Depending on the energy of the 1Se state relative to vacuum, strategies to generate the intraband PL should differ from material to material. This is because, in principle, the highest occupied electronic states vary depending on the material. For example, in case of the CdSe CQDs, a long-lived electron can be created when using multiphoton excitation in the presence of a hole quencher, whereas the hole quencher is unnecessary for the self-doped CQDs to generate the intraband PL.22–24,27,28,34,39,40,51,54,63,64,69 This is because the electrons of CdSe are short-lived due to the strong electron–hole Coulomb interaction. To avoid the electron–hole recombination, an efficient hole quencher is required. The multiphoton excitation method, in principle, can be extended to a wide variety of nanocrystals in order to generate the intraband PL spectra, although the signal-to-noise ratio must be improved.
Concluding remarks
In sum, a significant step forward has been possible in the studies on intraband transition through the self-doping of nanocrystals, providing fundamental information on charge carrier dynamics, and opening up a pathway to exploit the quantum confined states of colloidal nanocrystals beyond the bandgap transition. As opposed to previous doping methods requiring post-doping process, the self-doping method maintains carriers in the electronic states of the nanocrystals, making it possible to observe intraband transitions in steady state and ambient condition. The recently demonstrated self-doping approach provides a means to initiate a new direction for colloidal quantum dot research. To note, due to the high carrier density, the self-doped nanocrystal is very sensitive to the surface conditions. By altering the electrostatic potential of the surface which varies the direction of the surface dipole, the intraband transition can be quenched and recovered. The surface sensitivity may serve as useful properties for electronic, optoelectronic, sensor and photovoltaic applications. Interesting studies on the magnetic properties of the self-doped nanocrystal may open the possibility to harness the colloid nanocrystal for low-cost spintronics, non-volatile memory, infrared optoelectronics, catalyst, imaging, and quantum computing. In addition, by changing the metal proportion in CdxHg1−xSe alloy nanocrystal, a wide optical window ranging from the visible to mid-IR regime was obtained. As a nontoxic alternative to mid-IR intraband transition materials, Ag2Se colloidal quantum dots hold great promise. Lastly, the intraband PL in the mid-IR regime available with the low-cost semiconductor nanocrystals is promising for free space communications, medical imaging, and photocatalytic applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT, & Future Planning (NRF-2016R1C1B2013416).Notes and references
- J. P. Zimmer, S. Kim, S. Ohnishi, E. Tanaka, J. V. Frangioni and M. G. Bawendi, J. Am. Chem. Soc., 2006, 128, 2526–2527 CrossRef PubMed.
- S. Kim and M. G. Bawendi, J. Am. Chem. Soc., 2003, 125, 14652–14653 CrossRef PubMed.
- L. Sun, J. J. Choi, D. Stachnik, A. C. Bartnik, D. R. Hyun, G. G. Malliaras, T. Hanrath and F. W. Wise, Nat. Nanotechnol., 2012, 7, 369 CrossRef PubMed.
- G. J. Suparn, K. W. Song, G. W. Hwang, R. E. Correa, J. Scherer, E. A. Dauler, Y. Shirasaki, M. G. Bawendi and V. Bulovic, Adv. Mater., 2015, 27, 1437–1442 CrossRef PubMed.
- P. Guyot-Sionnest and J. A. Roberts, Appl. Phys. Lett., 2015, 107, 253104 CrossRef.
- D. V. Talapin, J. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389–458 CrossRef PubMed.
- G. H. Carey, A. L. Abdelhady, Z. Ning, S. M. Thon, O. M. Bakr and E. H. Sargent, Chem. Rev., 2015, 115, 12732–12763 CrossRef PubMed.
- M. Yuan, M. Liu and E. H. Sargent, Nat. Energy, 2016, 1, 16016 CrossRef.
- A. P. Alivisatos, Science, 1996, 271, 933–937 CrossRef.
- M. V. Kovalenko, L. Manna, A. Cabot, Z. Hens, D. V. Talapin, C. R. Kagan, V. I. Klimov, A. L. Rogach, P. Reiss, D. J. Milliron, P. Guyot-Sionnnest, P. G. Konstantatos, W. J. Parak, T. Hyeon, T. B. A. Korgel, C. B. Murray and W. Heiss, ACS Nano, 2015, 9, 1012–1057 CrossRef PubMed.
- S. V. Kershaw, A. S. Susha and A. L. Rogach, Chem. Soc. Rev., 2013, 42, 3033–3087 RSC.
- J. Y. Kim, O. Voznyy, D. Zhitomirsky and E. H. Sargent, Adv. Mater., 2013, 25, 4986–5010 CrossRef PubMed.
- S. A. McDonald, G. Konstantatos, S. Zhang, P. W. Cyr, E. J. Klem, L. Levina and E. H. Sargent, Nat. Mater., 2005, 4, 138–142 CrossRef PubMed.
- Z. Ning, O. Voznyy, J. Pan, S. Hoogland, V. Adinolfi, J. Xu, M. Li, A. R. Kirmani, J. P. Sun, J. Minor, K. W. Kemp, H. Dong, L. Rollny, A. Labelle, G. Carey, B. Sutherland, I. Hill, A. Amassian, H. Liu, J. Tang, O. M. Bakr and E. H. Sargent, Nat. Mater., 2014, 13, 822–828 CrossRef PubMed.
- A. Pandey and P. Guyot-Sionnest, J. Phys. Chem. Lett., 2010, 1, 45–47 CrossRef.
- J. M. Pietryga, Y. S. Park, J. Lim, A. F. Fidler, W. K. Bae, S. Brovelli and V. I. Klimov, Chem. Rev., 2016, 116, 10513–10622 CrossRef PubMed.
- M. Shim and P. Guyot-Sionnest, Nature, 2000, 407, 981–983 CrossRef PubMed.
- J. M. Harbold, H. Du, T. D. Krauss, K. S. Cho, C. B. Murray and F. W. Wise, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 1–6 CrossRef.
- C. Wang, M. Shim and P. Guyot-Sionnest, Science, 2001, 291, 2390–2392 CrossRef PubMed.
- J. D. Rinehart, A. M. Schimpf, A. L. Weaver, A. W. Cohn and D. R. Gamelin, J. Am. Chem. Soc., 2013, 135, 18782–18785 CrossRef PubMed.
- Z. Ning, O. Voznyy, J. Pan, S. Hoogland, V. Adinolfi, J. Xu, M. Li, A. R. Kirmani, J. Sun, J. Minor, K. W. Kemp, H. Dong, L. Rollny, A. Labelle, F. Carey, B. Sutherland, I. Hill, S. Amassian, H. Liu, J. Tang, O. M. Bakr and E. H. Sargent, Nat. Mater., 2014, 13, 4–10 CrossRef PubMed.
- P. Guyot-Sionnest and M. A. Hines, Appl. Phys. Lett., 1998, 72, 686–688 CrossRef.
- V. I. Klimov, A. A. Mikhailovsky, D. W. McBranch, C. A. Leatherdale and M. G. Bawendi, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, R13349–R13352 CrossRef.
- B. Yoon, J. Jeong and K. S. Jeong, J. Phys. Chem. C, 2016, 120, 22062–22068 CrossRef.
- P. Guyot-Sionnest, M. Shim, C. Matranga and M. Hines, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, R2181–R2184 CrossRef.
- V. I. Klimov and D. W. McBranch, Phys. Rev. Lett., 1998, 80, 4028–4031 CrossRef.
- A. Pandey and P. Guyot-Sionnest, Science, 2008, 322, 929–932 CrossRef PubMed.
- K. S. Jeong and P. Guyot-Sionnest, ACS Nano, 2016, 10, 2225–2231 CrossRef PubMed.
- B. L. Wehrenberg and P. Guyot-Sionnest, J. Am. Chem. Soc., 2003, 125, 7806–7807 CrossRef PubMed.
- D. Yu, C. Wang and P. Guyot-Sionnest, Science, 2003, 300, 1277–1280 CrossRef PubMed.
- Z. Deng, K. S. Jeong and P. Guyot-Sionnest, ACS Nano, 2014, 8, 11707–11714 CrossRef PubMed.
- E. Lhuillier, M. Scarafagio, P. Hease, B. Nadal, H. Aubin, X. Z. Xu, N. Lequeux, G. Patriarche, S. Ithurria and B. Dubertret, Nano Lett., 2016, 16, 1282–1286 CrossRef PubMed.
- S. Brovelli, W. K. Bae, F. Meinardi, B. Santiago Gonzalez, M. Lorenzon, C. Galland and V. I. Klimov, Nano Lett., 2014, 14, 3855–3863 CrossRef PubMed.
- H. Liu, S. Keuleyan and P. Guyot-Sionnest, J. Phys. Chem. C, 2012, 116, 1344–1349 CrossRef.
- H. Gerischer, J. Electroanal. Chem., 1977, 82, 133–143 CrossRef.
- C. K. Chiang, M. A. Druy, S. C. Gaul, A. J. Heeger, E. J. Louis, A. G. MacDiarmid, Y. W. Park and H. Shirakawa, J. Am. Chem. Soc., 1978, 100, 1013–1015 CrossRef.
- W. K. Koh, A. Y. Koposov, J. T. Stewart, B. N. Pal, I. Robel, J. M. Pietryga and V. I. Klimov, Sci. Rep., 2013, 3, 2004 CrossRef PubMed.
- L. M. Peter, Chem. Rev., 1990, 90, 753–769 CrossRef.
- K. S. Jeong, Z. Deng, S. Keuleyan, H. Liu and P. Guyot-Sionnest, J. Phys. Chem. Lett., 2014, 5, 1139–1143 CrossRef PubMed.
- J. Jeong, B. Yoon, Y.-W. Kwon, D. Choi and K. S. Jeong, Nano Lett., 2017, 17, 1187–1193 CrossRef PubMed.
- D. A. Schwartz, N. S. Norberg, Q. P. Nguyen, J. M. Parker and D. R. Gamelin, J. Am. Chem. Soc., 2003, 125, 13205–13218 CrossRef PubMed.
- M. Freitag, H. Y. Chiu, M. Steiner, V. Perebeinos and P. Avouris, Nat. Nanotechnol., 2010, 5, 497–501 CrossRef PubMed.
- L. Tang, R. Ji, X. Li, G. Bai, C. P. Liu, J. Hao, J. Lin, H. Jiang, K. S. Teng, Z. Yang and S. P. Lau, ACS Nano, 2014, 8, 6312–6320 CrossRef PubMed.
- L. M. Lawton, N. H. Mahlmeister, I. J. Luxmoore and G. R. Nash, AIP Adv., 2014, 4, 087139 CrossRef.
- J. W. Stouwdam and F. C. J. M. Van Veggel, Nano Lett., 2002, 2, 733–737 CrossRef.
- C. M. Andolina, S. E. Crawford, A. M. Smith, K. A. Johnston, P. J. Straney, L. E. Marbella, N. L. Tolman, T. J. Hochuli and J. E. Millstone, ChemNanoMat, 2018, 4, 265–268 CrossRef.
- J. L. Liu, Y. T. Yang, C. Te Lin, Y. J. Yu, J. K. Chen and L. K. Chu, J. Phys. Chem. C, 2017, 121, 878–885 CrossRef.
- A. Narita, I. A. Verzhbitskiy, W. Frederickx, K. S. Mali, S. A. Jensen, M. R. Hansen, M. Bonn, S. D. Feyter, C. Casiraghi, X. Feng and K. Müllen, ACS Nano, 2014, 8, 11622–11630 CrossRef PubMed.
- T. R. Jensen, M. D. Malinsky, C. L. Haynes and R. P. Van Duyne, J. Phys. Chem. B, 2000, 104, 10549–10556 CrossRef.
- X. Ye, L. Jin, H. Caglayan, J. Chen, G. Xing, C. Zheng, V. D. Nguyen, Y. Kang, N. Engheta, C. R. Kangan and C. B. Murray, ACS Nano, 2012, 6, 2804–2817 CrossRef PubMed.
- A. Robin, C. Livache, S. Ithurria, E. Lacaze, B. Dubertret and E. Lhuillier, ACS Appl. Mater. Interfaces, 2016, 8, 27122–27128 CrossRef PubMed.
- K. A. Higginson, M. Kuno, J. Bonevich, S. B. Qadri, M. Yousuf and H. Mattoussi, J. Phys. Chem. B, 2002, 106, 9982–9985 CrossRef.
- S. Keuleyan, E. Lhuillier and P. Guyot-Sionnest, J. Am. Chem. Soc., 2011, 133, 16422–16424 CrossRef PubMed.
- E. Lhuillier and P. Guyot-Sionnest, IEEE J. Sel. Top. Quantum Electron., 2017, 23, 6000208 Search PubMed.
- I. H. Campbell, S. Rubin, T. A. Zawodzinski, J. D. Kress, R. L. Martin, D. L. Smith, N. N. Barashkov and J. P. Ferraris, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 14321–14324 CrossRef.
- P. R. Brown, D. Kim, R. R. Lunt, N. Zhao, M. G. Bawendi, J. C. Grossman and V. Bulovic, ACS Nano, 2014, 8, 5863–5872 CrossRef PubMed.
- H. Wang, I. Barcelo, T. Lana-Villarreal, R. Gómez, M. Bonn and E. Cánovas, Nano Lett., 2014, 14, 5780–5786 CrossRef PubMed.
- J. Y. Woo, J. H. Ko, J. H. Song, K. Kim, H. Choi, Y. H. Kim, D. C. Lee and S. Jeong, J. Am. Chem. Soc., 2014, 136, 8883–8886 CrossRef PubMed.
- I. Moreels, K. Lambert, D. De Muynck, F. VAnhaecke, D. Poelman, J. C. Martins, G. Allan and Z. Hens, Chem. Mater., 2007, 19, 6101–6106 CrossRef.
- I. Moreels, K. Lambert, D. Smeets, D. De Muynck, T. Nollet, J. C. Martins, F. Vanhaecke, A. Vantomme, C. Delerue, G. Allan and Z. Hens, ACS Nano, 2009, 3, 3023–3030 CrossRef PubMed.
- H. Liu and P. Guyot-Sionnest, J. Phys. Chem. C, 2015, 119, 14797–14804 CrossRef.
- D. J. Norris, A. L. Efros and S. C. Erwin, Science, 2008, 319, 1776–1779 CrossRef PubMed.
- D. Choi, B. Yoon, D.-K. Kim, H. Baik, J.-H. Choi and K. S. Jeong, Chem. Mater., 2017, 29, 8548–8554 CrossRef.
- J. Kim, B. Yoon, J. Kim, Y. Choi, Y.-W. Kwon, S. K. Park and K. S. Jeong, RSC Adv., 2017, 7, 38166–38170 RSC.
- A. Agrawal, S. H. Cho, O. Zandi, S. Ghosh, R. W. Johns and D. J. Milliron, Chem. Rev., 2018, 118, 3121–3207 CrossRef PubMed.
- A. M. Schimpf, N. Thakkar, C. E. Gunthardt, D. J. Masiello and D. R. Gamelin, ACS Nano, 2013, 8, 1065–1072 CrossRef PubMed.
- F. Scotognella, G. D. Valle, A. R. S. Kandada, M. Zavelani-Rossi, A. Comin, S. Longhi, L. Manna, G. Lanzani and F. Tassone, Eur. Phys. J. B, 2013, 86, 154 CrossRef.
- G. Shen and P. Guyot-Sionnest, J. Phys. Chem. C, 2016, 120, 11744–11753 CrossRef.
- M. Park, D. Choi, Y. Choi, H. B. Shin and K. S. Jeong, ACS Photonics, 2018, 5, 1907–1911 CrossRef.
- A. Sahu, L. Qi, M. S. Kang, D. Deng and D. J. Norris, J. Am. Chem. Soc., 2011, 133, 6509–6512 CrossRef PubMed.
- A. Sahu, A. Khare, D. D. Deng and D. J. Norris, Chem. Commun., 2012, 48, 5458–5460 RSC.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |