
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSecondary nucleation in amyloid formation
Mattias
Törnquist
 a,
Thomas C. T.
Michaels
bc,
Kalyani
Sanagavarapu
a,
Xiaoting
Yang
a,
Georg
Meisl
b,
Samuel I. A.
Cohen
b,
Tuomas P. J.
Knowles
bd and
Sara
Linse
*a
a,
Thomas C. T.
Michaels
bc,
Kalyani
Sanagavarapu
a,
Xiaoting
Yang
a,
Georg
Meisl
b,
Samuel I. A.
Cohen
b,
Tuomas P. J.
Knowles
bd and
Sara
Linse
*a
aLund University, Department of Biochemistry and Structural Biology, Chemical Centre, PO Box 124, SE221 00 Lund, Sweden. E-mail: sara.linse@biochemistry.lu.se
bCambridge University, Department of Chemistry, Lensfiled Road, Cambridge, UK
cPaulson School of Engineering and Applied Sciences, Harvard University, Cambridge, MA 02138, USA
dCambridge University, Cavendish Laboratory, Department of Physics, JJ Thomson Avenue, Cambridge, UK
First published on 6th July 2018
Abstract
Nucleation of new peptide and protein aggregates on the surfaces of amyloid fibrils of the same peptide or protein has emerged in the past two decades as a major pathway for both the generation of molecular species responsible for cellular toxicity and for the autocatalytic proliferation of peptide and protein aggregates. A key question in current research is the molecular mechanism and driving forces governing such processes, known as secondary nucleation. In this context, the analogies with other self-assembling systems for which monomer-dependent secondary nucleation has been studied for more than a century provide a valuable source of inspiration. Here, we present a short overview of this background and then review recent results regarding secondary nucleation of amyloid-forming peptides and proteins, focusing in particular on the amyloid β peptide (Aβ) from Alzheimer's disease, with some examples regarding α-synuclein from Parkinson's disease. Monomer-dependent secondary nucleation of Aβ was discovered using a combination of kinetic experiments, global analysis, seeding experiments and selective isotope-enrichment, which pinpoint the monomer as the origin of new aggregates in a fibril-catalyzed reaction. Insights into driving forces are gained from variations of solution conditions, temperature and peptide sequence. Selective inhibition of secondary nucleation is explored as an effective means to limit oligomer production and toxicity. We also review experiments aimed at finding interaction partners of oligomers generated by secondary nucleation in an ongoing aggregation process. At the end of this feature article we bring forward outstanding questions and testable mechanistic hypotheses regarding monomer-dependent secondary nucleation in amyloid formation.
1. Introduction
Monomer-dependent secondary nucleation has been identified during the 1900s and 2000s as a key step in the formation of self-assembled aggregates of monomers covering a wide range of chemical space. This includes the conversion of monomeric peptides and proteins to large fibrillar aggregates known as amyloid fibrils associated with a range of human disorder (Fig. 1).3–7 The devastating nature of these debilitating diseases, including Alzheimer's, Parkinson's and Huntington's diseases, and their increasing prevalence, has stimulated a major research interest in finding the molecular basis of amyloid diseases. Despite massive effort, the sheer complexity of the systems involved has hampered detailed molecular understanding and effective treatment for the diseases in question.8 Much of this complexity goes beyond the scope of this feature article, yet we address here one aspect of the puzzle, which involves the molecular mechanisms for amyloid formation. Studies in this area aim to identify the microscopic steps behind the overall reaction pathway. These mechanistic studies and findings provide the basis for the identification of which of the underlying steps are associated with the emergence of toxicity to human cells and tissues, and screening for inhibitors that can act specifically on those steps with an aim to limit toxicity.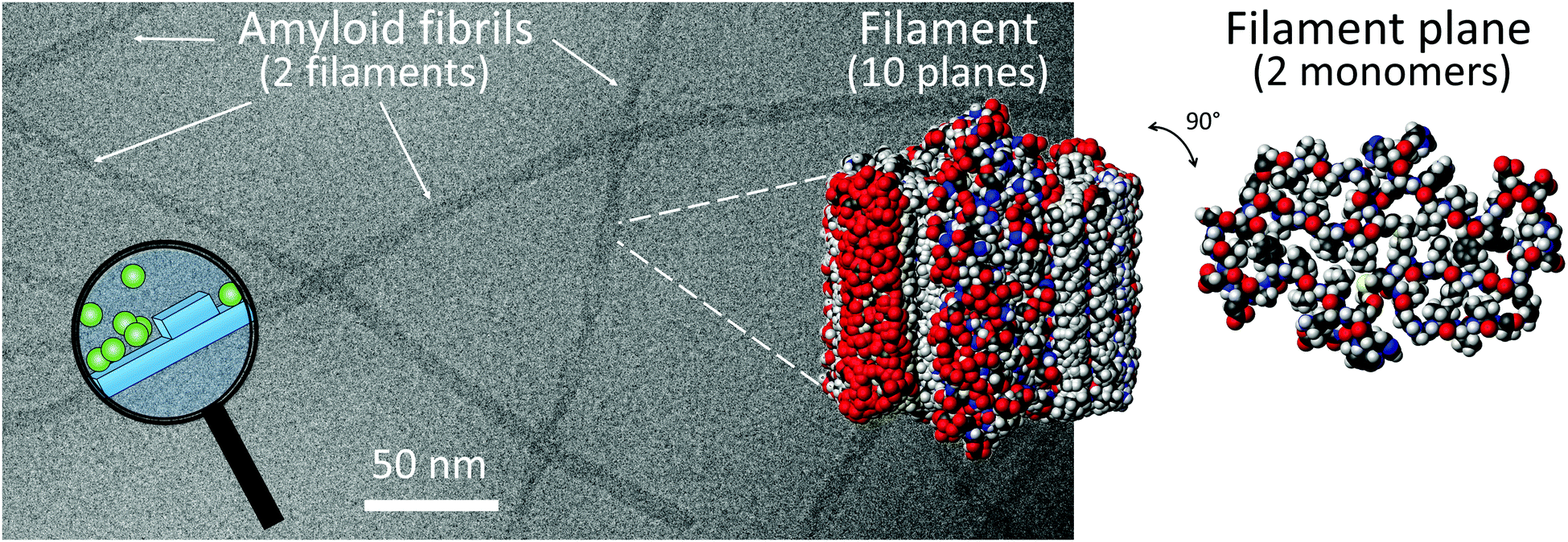 | ||
| Fig. 1 A Cryo-EM image of Aβ42 fibrils composed of two filaments. CPK models of 10 planes of a filament and one plane of a filament with two monomers, prepared using pdb file 5KK31 and Molmol.2 The magnifying glass highlights a cartoon over monomer-dependent secondary nucleation on the fibril surface. | ||
Amyloid fibrils of a wide range of peptides and proteins display similar structures irrespective of their sequence,3–6 in stark contrast to the wide variations seen in the folded structures and functions of the native states of proteins which are largely governed by their amino acid sequences.9 It has even been postulated that amyloid may represent a generic structure that can be formed by peptides and protein of any sequence, at least under appropriate solution conditions.10 Amyloid fibrils display a highly repetitive packing of multiple identical protein chains in extended β-sheets.11 The densely packed and highly ordered cores of amyloid fibrils have been revealed by a number of high resolution models based on data from X-ray diffraction, X-ray crystallography, solid state NMR spectroscopy and cryo electron microscopy.1,12–20 The sensitivity of the formed fibril structure to the solution conditions was recently illustrated for Aβ42. While two virtually identical models of Aβ42 fibrils were presented based on independent sample preparation and ssNMR studies at close to physiological pH and ionic strength1,16 a very different model was inferred based on ssNMR and cryo-EM data obtained for fibrils formed at acidic pH in high concentration of acetonitrile.20
Amyloids are thus self-assembled structures of multiple identical monomers. In chemistry, materials science and biology there are many other examples where the emergence of structure and function relies on the self-assembly of monomeric building blocks into extended structures of more or less defined size in one, two or three dimensions, e.g. hydrogels,21,22 nanoparticles,23 nanowires,24 zeolites,25 nano-capsules,26 drug crystals,27 protein crystals,28 virus coats,29 cytoskeletal filaments,30 liposomes,31 protein fibres in the context of natural and artificial nanomaterials32–34 and implicated in the aetiology of human diseases.6,7 The repetition of the bonding pattern between monomers and the chirality of these interactions controls the structural order and symmetry on much larger length scales.35 The surface properties of the aggregates govern their interactions with other molecules and aggregates, and lead to more or less organized aggregate networks or bundles, which in turn may promote specific materials properties of synthetic or natural self-assembled systems.
This Feature Article aims to provide an overview of some recent advances regarding our understanding of the self-assembly mechanisms in amyloid systems, in particular with respect to the secondary nucleation of monomers on the surfaces of amyloid fibrils. We argue that insights from many decades of studies of a wide range of other self-assembled systems may provide clues that can forward our understanding of amyloid formation mechanisms, and we will therefore include a brief summary of some results from such work.
2. Self-assembly through nucleation and growth
The formation of molecular aggregates involves the transition of a system initially in a homogeneous solution phase, to form a new aggregated phase co-existing with a monomer solution phase. Such transitions can in principle occur either through spinodal decomposition, which involves the spontaneous coalescence of a large number of molecules initially in homogeneous solution as monomers, or through nucleation and growth. The simultaneous coalescence of thousands or more molecules into an ordered aggregate is unlikely, and instead, self-assembled protein filaments typically emerge through nucleation-and-growth processes,36 where the formation of nuclei is an obligatory step and bottle-neck in the overall reaction towards a new phase or a new structure with lower free energy. In classical nucleation theory, very small clusters of monomers have a high interfacial energy towards water and hence a high free energy per monomer; as a consequence, they have a high probability to dissociate into their component monomers. With increasing size, however, stabilization from monomer–monomer interactions within the cluster becomes more significant, and the probability of a nucleus to grow through monomer addition rather than shrink through dissociation increases. Critical nuclei are commonly defined as the smallest aggregates in the process that are stable enough to go on to form the new phase with a higher probability than to dissolve. In energetic terms, critical nuclei are thus the species with highest Gibbs free energy along the aggregation pathway. Nucleation is an intrinsically slow event characterized by a high free energy barrier, whereas additional growth occurs at higher rate with a lower free energy barrier. Nucleation reactions may in general follow complicated pathways that populate a cascade of intermolecular metastable species37 or pre-nucleation clusters.Coarse-grained simulations of amyloid formation have revealed that non-specific (non-fibrillar) contacts between peptides promote the formation of relatively disordered clusters, which undergo a structural conversion to the fibrillar state.38–40 For some systems, the nucleation step may be preceded by liquid–liquid phase separation,41 leading to the formation of liquid droplets of a dense liquid phase which is a precursor for solid nuclei to form. Interestingly, liquid/liquid phase separation behaviour of this type may be implicated in the nucleation of amyloid and amyloid-like fibrils.42–44 Thus, the nucleation of amyloid fibrils follows many but not all aspects of classical nucleation theory, and involves at least an additional slow conformational conversion step as discussed in Sections 3.7 and 4.
2.1 Solubility, stability, metastability, saturation and supersaturation
Self-assembled structures emerge through the spontaneous association of monomers. Homogeneous monomer solutions may be classified as stable, metastable or unstable (Fig. 2) depending on the monomer concentration at the specified temperature and solution conditions.45–49 Monomer solutions are stable up to a solubility limit, S0, also called the crystalline solubility. At concentrations below S0, the solution remains monomeric for infinite time and the thermodynamically most stable state of the system is a homogeneous solution of monomers. Under such conditions, the Gibbs free energy of transferring a monomer from the solution phase to the aggregated phase is positive and thus thermodynamically unfavourable. Above S0, the solution is supersaturated. Such situation may arise after a sudden change in temperature or pH, or by dilution into aqueous solution from an organic solvent. Under supersaturated conditions, the most stable state is a two-phase system, with aggregated structures co-existing with the monomer. The system remains metastable, and as such only rare fluctuations are significant enough to lead to the formation of a new aggregated phase within the soluble phase through nucleation and growth. However, even though nucleation is largely suppressed, already formed aggregates grow until the monomer concentration reaches S0 and equilibrium is reached. In the unstable regime, the monomer concentration is high enough such that the homogeneous phase is unstable with respect to even small density fluctuations and spontaneously decomposes into a two phase system. The width of the unstable and metastable zones is time dependent48 since for longer times, the probability of observing larger fluctuations leading to nucleation in the metastable zone increases and as such metastable solutions with high concentration display nucleation and growth more rapidly than metastable solutions with lower monomer concentration.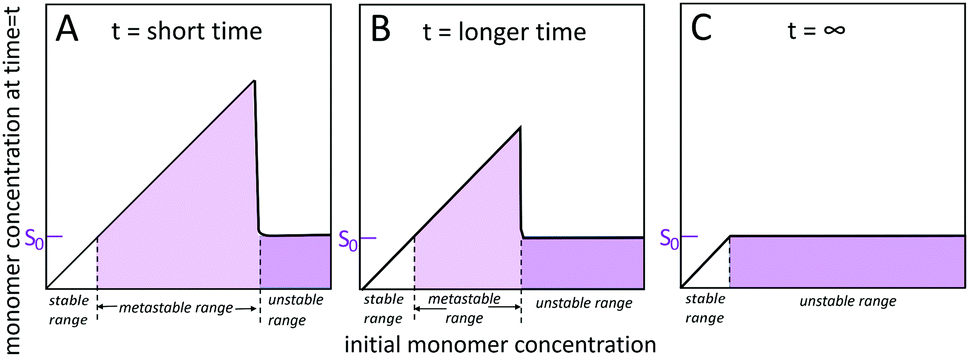 | ||
| Fig. 2 Solubility and monomer concentration in self-assembling systems. S0 is the monomer concentration that will exist in equilibrium with aggregates after a system with total concentration > S0 has reached equilibrium. The lower limit of the metastable zone (light purple) is always S0, but its upper limit will be lower the longer the incubation time of the samples (A and B) and will eventually disappear (C). | ||
2.2 Definition of primary nucleation
Primary nucleation is a reaction involving monomers (Fig. 3) without contributions from already formed aggregates. In this process, monomers associate into small aggregates that can reach sizes at which they growth faster by monomer addition compared to the rate by which they dissociate. Primary nucleation may occur in bulk solution (homogeneous nucleation) or at a surface (heterogeneous nucleation). This surface could for example be the wall of a reaction vessel, the surface of an assembly of another protein or some other substance such as a lipid vesicle,50 or the air–water interface.51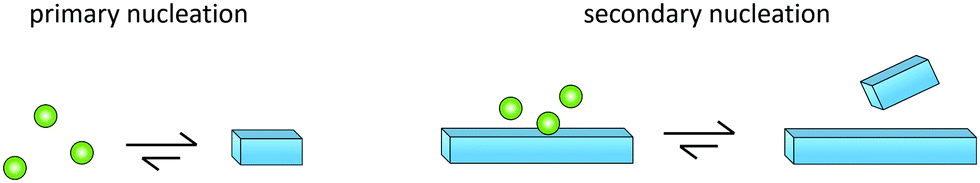 | ||
| Fig. 3 Cartoon of primary and secondary nucleation reactions. Monomers of one substance may nucleate in solution (primary nucleation, left) or on the surface of an already existing aggregate (secondary nucleation, right). Green and blue colour symbolises the conformation in free state and aggregated state, respectively. | ||
2.3 Definition of secondary nucleation
Monomer-dependent secondary nucleation is defined as a process whereby nucleus formation from monomers is catalysed by existing aggregates composed of the same type of monomeric building blocks. Usually this takes the form of monomers forming a nucleus on the surface of an already existing aggregate (Fig. 3). Secondary nucleation thus happens in the presence of a parent seed aggregate. Secondary nucleation is distinct from heterogeneous primary nucleation in that no foreign surface is involved and while the molecular mechanisms of the two processes have some similarities, the resulting overall kinetics of aggregation are quite distinct. Here we will briefly review some general features and historical findings regarding secondary nucleation in self-assembly reactions in general, before we highlight findings and hypotheses regarding secondary nucleation in amyloid formation.2.4 Occurrence of secondary nucleation in non-amyloid systems
The autocatalytic amplification of aggregate mass due to secondary nucleation has been observed in self-assembly reactions for more than a century.42,52,53 Examples of 3D assemblies for which secondary nucleation leads to propagation of aggregates can be found in the crystallization of proteins and small molecules,54–57 and even the formation of ice crystals can be governed by secondary nucleation.58 Other 3D assembled systems for which secondary nucleation has been reported are CaF and MgF particles,59 silver nanoparticles,60 zeolites,61–63 and other inorganic and organic compounds,53,64–66 as well as phase transitions in superfluid helium.67 Remarkably, molecular dynamics simulations have revealed secondary nucleation in crystallization of a simple model system – that of Lennard-Jones particles – depending on the level of super-saturation of the system.64 Examples of assemblies of low dimensionality are actin filaments and the fibrous aggregates of sickle cell haemoglobin.68–702.5 Mechanisms of secondary nucleation inferred from non-amyloid systems
Forces between monomer clusters and a crystal surface favour secondary nucleation. The interfacial energy, the molar volume of the solid phase and the difference in chemical potential between the solution and solid phase have been noted as key parameters governing the rate of secondary nucleation.71,72 Similar to primary nucleation, the rate of secondary nucleation increases abruptly above a certain level of super-saturation, however the level of supersaturation at which this occurs can be lower than for primary nucleation (Fig. 4).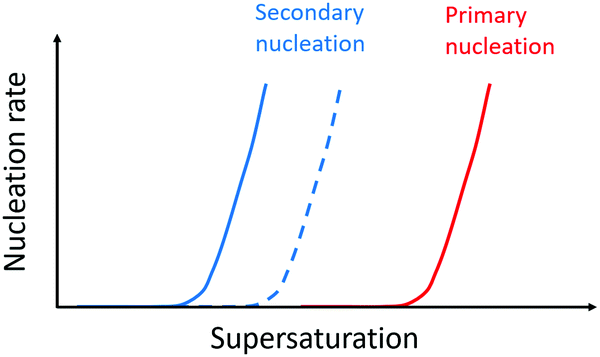 | ||
| Fig. 4 Nucleation rate as a function of supersaturation. Primary nucleation rate (red) and secondary nucleation rate for monomers of the same enantiomer as the monomers in the aggregate (blue solid line) and the surface nucleation rate for monomers of the opposite enantiomer relative to the monomers in the aggregate (dashed blue line). Adapted from ref. 72. | ||
Seeding of a supersaturated monomer solution with pre-formed crystals leads to the generation of new crystals with the same morphology, chirality, crystal packing and space group as the seed.54 This fact is exploited in industrial crystallizers where care is taken to design rectors and solution conditions such that secondary nucleation can be utilized as a route to more homogeneous product.73 In mixtures of enantiomers, a higher rate of secondary nucleation has been observed for the monomers in solution which are of the same enantiomer as those in the aggregate, because there is a concentration window in terms of supersaturation where the secondary nucleation of the same enantiomer is fast while the surface-catalysed nucleation of the opposite enantiomer is negligible (Fig. 4). Thus, in many cases, secondary nucleation has been observed to be enantioselective.54,72
Several molecular mechanism of monomer-dependent secondary nucleation have been discussed. In one mechanism, clusters of monomers form in a supersaturated solution of monomers. If a crystal seed is added to such a solution, a large number of clusters may form in the vicinity of the crystal surface, where they may coalesce and form nuclei.71,74 In this mechanism, dissociation of the formed nuclei from the parent surface to the bulk is an essential step to complete the secondary nucleation process, and this detachment step may be enhanced by shear flow or collisions. Contact nucleation is used in the literature as a term to denote secondary nucleation that occurs as a consequence of collisions but with no visible impact on the parent crystal, thus the origin of the nucleus is from the monomers in the solution phase. Secondary nucleation has been associated with the appearance of dendrite-like irregularities on the surface of the parent crystal, the detachment of which seems to rely on coarsening of these structures at the remote end concomitant with thinning at the end closest to the parent crystal (ref. 75 and references therein).
3. Secondary nucleation in amyloid formation
In the following we will review some recent results regarding secondary nucleation in amyloid formation from our own and other investigators’ work. Based on these results we will put forward some open questions and testable hypotheses regarding mechanisms of secondary nucleation and discuss how these questions might be addressed in future experiments.3.1 Monomer-dependent secondary nucleation is observed in amyloid formation from several peptides and proteins
Historically, monomer-dependent secondary nucleation in the formation of protein filaments was first identified in the 1980's in the context of pioneering studies on the aggregation of the mutant haemoglobin HbS,68–70 a process linked to sickle-cell anaemia. Later, monomer-dependent secondary nucleation in amyloid formation has been inferred for the aggregation of several other proteins, including, for example insulin,76 Islet Amyloid Poly Peptide (IAPP),77 amyloid-β peptide (Aβ),78,79 α-synuclein (α-syn)80,81 and carbonic anhydrase.82 These proteins and aggregation processes have been implicated in devastating diseases such as diabetes (Insulin, IAPP), Alzheimer's (Aβ,) and Parkinson's (α-syn) diseases. In all these systems, proliferation of aggregates and the fact that once initiated the disease is very difficult to cure, may be related to the autocatalytic amplification of aggregate mass due to secondary nucleation (Fig. 5).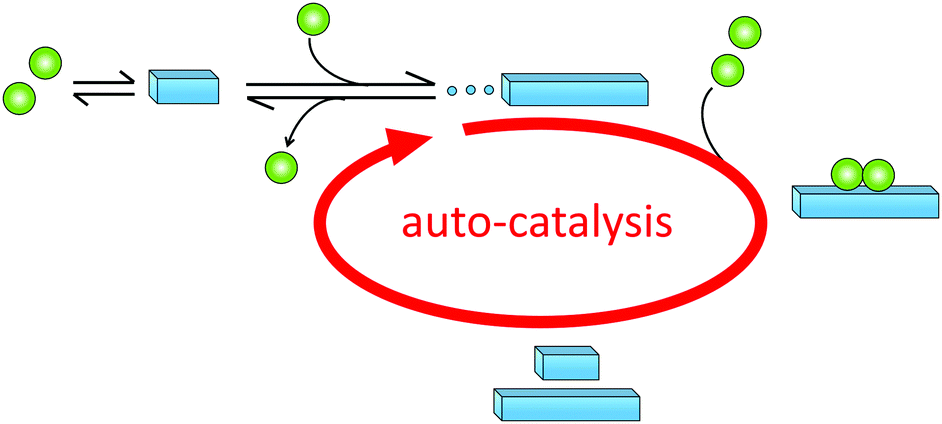 | ||
| Fig. 5 Auto-catalytic amplification of aggregate mass due to monomer-dependent secondary nucleation. Monomers (green or blue) may undergo primary nucleation (left). Aggregates may elongate by monomer addition (middle) or catalyse the generation of new aggregates through monomer-dependent secondary nucleation (right, auto-catalytic cycle). | ||
3.2 Discovery of monomer-dependent secondary nucleation in amyloid formation of Aβ42
In the case of Aβ, monomer-dependent secondary nucleation was discovered as late as during the current decade78 based on three levels of evidence, as follows:(1) The first indication came from the fact that experimental data for the concentration-dependent time course of amyloid fibril formation from a solution of Aβ42 monomers could not be explained by classical models of nucleated polymerization involving primary nucleation and fibril growth (Fig. 6A).78 In particular, the data shows a lag phase followed by sharp, exponential-type growth in the fibril mass, which was in contrast to the polynomial growth predicted in the absence of secondary nucleation pathways (Fig. 6A). Moreover, the concentration dependence of the data could not be described by the addition of a filament fragmentation step,83 which is an alternative auto-catalytic mechanism to monomer-dependent secondary nucleation that results in a weaker concentration dependence to that observed. However, the data could be reproduced to a high level of accuracy by a model including primary nucleation, fibril growth and secondary nucleation of monomers on the surface of fibrils (Fig. 6A).84–87 While this global analysis of the concentration-dependent data provided initial evidence of the role of secondary nucleation in Aβ42 aggregation, the analysis also provided further predictions for the outcome of new experiments that were then verified.
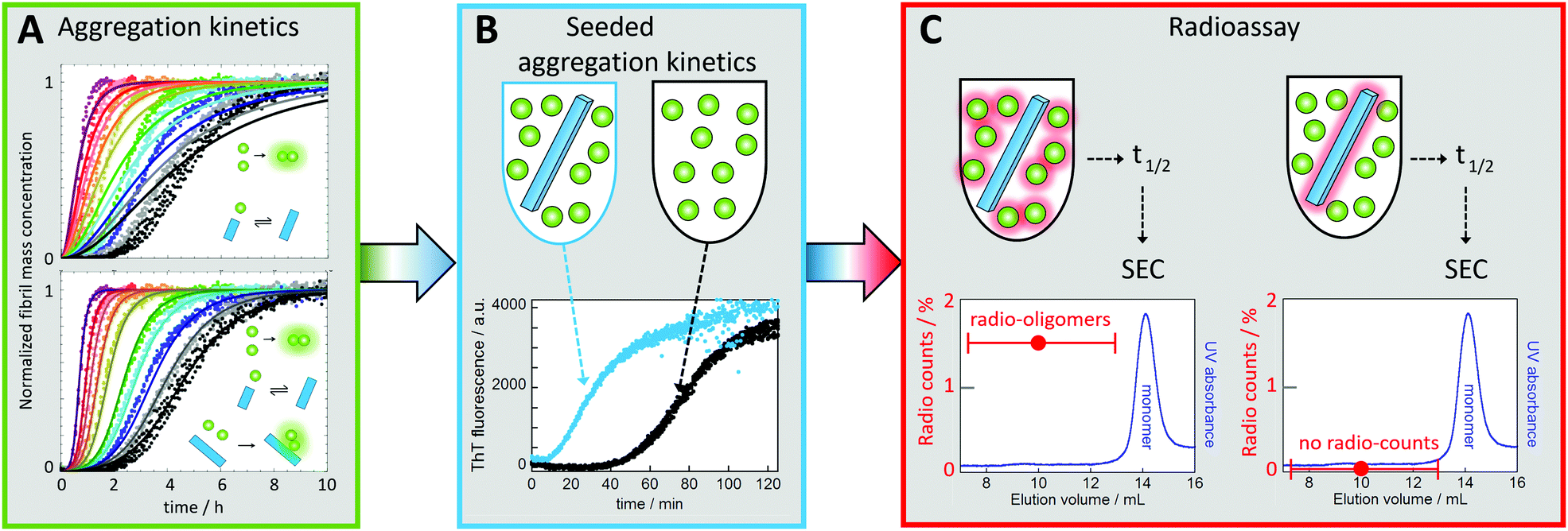 | ||
| Fig. 6 Discovery of monomer-dependent secondary nucleation of Aβ42. (A) Fibril formation kinetics starting from monomer solutions at several concentrations (individual colours, in quadruplicate). Top: Best fit for a model with primary nucleation and elongation. Bottom: Best fit when also secondary nucleation of monomers on aggregate surface is included. (B) Experiments in the presence of low concentration of seeds (blue) show reduced lag phase relative to non-seeded reaction (black) signifying a secondary process. (C) Radio-assay with radioactive monomer and 1% unlabeled seed (left sample) or unlabeled monomer and 1% radioactive seed (right sample). Both samples were incubated until half the monomers had converted to fibrils, which were removed by centrifugation. The supernatant was subjected to gel filtration to collect the oligomer fractions. Radio-oligomers were detected from the left sample but not for the right sample. This shows that new aggregates (oligomers) are generated from monomer in a reaction catalyzed by the seed fibrils.78 | ||
(2) One key prediction was that the addition of small amounts of preformed fibrils (seeds), of a quantity so small that the sigmoidal-like shape of the growth curve is conserved, would bypass the primary nucleation process, resulting in almost all new fibrils being generated through secondary nucleation. Indeed, it was observed that the addition of a small fraction of preformed seeds reduced the timescale of the aggregation reaction to such an extent that the entirety of the peptide was converted into aggregates before the corresponding reaction in the absence of seeds had proceeded through the lag phase (Fig. 6B).78 These data confirmed that the presence of fibrils at the start of the reaction resulted in a fibril-dependent nucleation process that did not occur in their absence.
(3) Finally, radio-isotope labelling was used to pinpoint the origin of the new aggregates being generated in a manner dependent on the presence of fibrils in the pre-seeded reaction. When radio-active monomer was mixed with unlabelled seeds, radioactive oligomers (up to ca. 20-mers) were found to be formed. By contrast, no radioactive oligomers were found when unlabelled monomer was mixed with radioactive seeds (Fig. 6C). These data showed conclusively that new aggregates are generated from monomer in a fibril-catalysed reaction, rather than being breakdown products due to fragmentation of the pre-formed seeds.78 Note that the reason dimers were excluded from the analysis was to avoid contamination by monomers, which are present at much higher concentration than the oligomers.
Thus while kinetic analysis indicated that the aggregation mechanism is dominated by a secondary pathway, seeded experiments and the use of specific isotope labelling identified unequivocally the secondary pathway as a process that produces new aggregates from monomers on the surface of fibrils.78
Interestingly, after establishing that Aβ42 aggregation is dominated by monomer-dependent secondary nucleation,78 the efficient shortening of the characteristic lag phase of Aβ42 amyloid formation in a manner dependent on seed concentration was utilized as a tool to quantify the fibril concentration as a function of time during the lag phase.88 It was found that Aβ42 fibrils can be detected from very early stages of the lag phase, that their concentration grows almost exponentially during the lag phase (or rather proportional to cosh(k·t) − 1)88 as also found for sickle cell haemoglobin,70 long before they can be detected by bulk techniques, the signal-to-noise of which may require on the order of 1% of the monomers converted to fibrils for detection to be possible.
3.3 Monomer-dependent secondary nucleation of α-syn
The α-syn protein, involved in Parkinson's disease, displays monomer-dependent secondary nucleation at mildly acidic pH.81 For this protein, primary nucleation in bulk solution is extremely slow. To be able to observe aggregation within a reasonable experimental time-frame under quiescent conditions, foreign surfaces need to be introduced, e.g. negatively charged phospholipid membranes50 or polystyrene surfaces in the form of sample containers89 or nanoparticles90 to promote heterogeneous primary nucleation. Alternatively, aggregation can be monitored for solutions of monomer that are supplemented with low concentration of preformed fibrils, which bypass the need for primary nucleation altogether.80,81 In the case of α-syn, the mechanism of fibril propagation and growth is dependent on pH. At neutral pH, the reaction is dominated by elongation of seeds, whereas at pH below 6.0, a secondary process is significant.80 The existence of secondary nucleation was thus first inferred from the requirement of a secondary process to produce reasonable global fits to aggregation data and the significant shortening of the lag phase upon addition of low (sub-%) concentrations of seeds.80 As described above for Aβ42, additional experiments were then needed to assess whether this secondary process can be ascribed to the nucleation of monomers on the surface of fibrils. A combination of trap-and-seed experiments, quartz crystal balance with dissipation and centrifugal sedimentation analyses of size distribution was used, and the data revealed that the secondary process observed at mildly acidic pH is indeed monomer-dependent secondary nucleation on the surface of fibrils.813.4 Thermodynamic signature
Mapping energy landscapes has proved to be a powerful tool for studying reaction mechanisms across many fields, and this approach has recently been extended to amyloid formation in a study of Aβ42 aggregation.91 The results revealed that monomer-dependent secondary nucleation of Aβ42 displays a distinct thermodynamic signature compared with primary nucleation, even though both processes generate aggregates from soluble peptides. Specifically, although the rate constant for both primary nucleation and fibril elongation increase at higher temperatures, the rate constant for secondary nucleation has only a very weak dependence on temperature and rather increases slightly with decreasing temperature (Fig. 7). While primary nucleation is a slow event with a high free energy barrier, secondary nucleation was found in the case of Aβ42 to be associated with a four-fold reduction in the free energy barrier relative to primary nucleation, demonstrating the remarkably effective manner in which amyloid fibrils catalyse the nucleation of new aggregates from monomers.91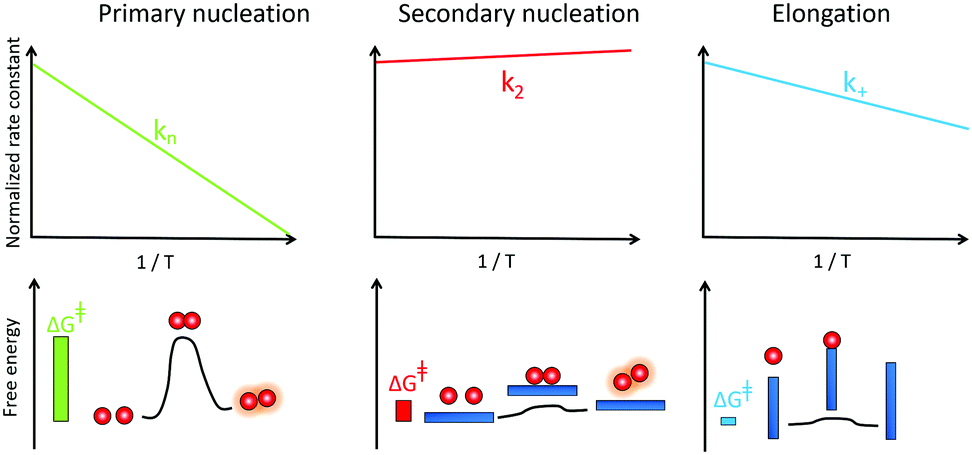 | ||
| Fig. 7 Temperature dependence of the rate constant for monomer-dependent secondary nucleation (middle) compared to primary nucleation (left) and elongation (right) and the derived barriers. | ||
Furthermore, the interactions between monomers and amyloid fibrils were found to not only change the rate constant characterising nucleation, but also to fundamentally reverse the thermodynamic signature of this process relative to primary nucleation.91 Specifically, the energy barriers for both primary nucleation and fibril elongation were found to be enthalpic in nature, with a favourable entropy of activation, indicating that the hydrophobic effect plays a dominant role in both processes. By contrast, the enthalpic barrier is abolished for secondary nucleation and the lowered free energy barrier was shown to be entirely entropic in nature. These results are analogous to surface catalyzed reactions involving an exothermic pre-binding step92 and reveal that the catalytic efficiency of Aβ42 fibril surfaces results from the enthalpic stabilisation of adsorbing peptides in conformations amenable to nucleation, driving a significant lowering of the activation energy barrier for nucleation.91
3.5 Underlying molecular events
Secondary nucleation of monomers on fibrils surface involves at least three molecular events: peptide arrival at the fibrils surface, product formation and release. In analogy with enzyme catalysis, secondary nucleation may saturate at high monomer (substrate) concentration (Fig. 8). For enzyme catalysis, saturation kinetics may be observed when there is a substrate-binding equilibrium preceding product formation and release. Aβ40 was the first case for which such saturation at high monomer concentration was observed,79 implying that secondary nucleation is not a single-step reaction. Saturating secondary nucleation has since been observed for Aβ42 upon reduced electrostatic repulsion due to salt screening,93,94 pH variation,95 or mutation.95–97 The composite steps may include association of monomer, or small clusters of monomers, to the surface of fibrillar aggregates, nucleation on the surface, and finally detachment (Fig. 8).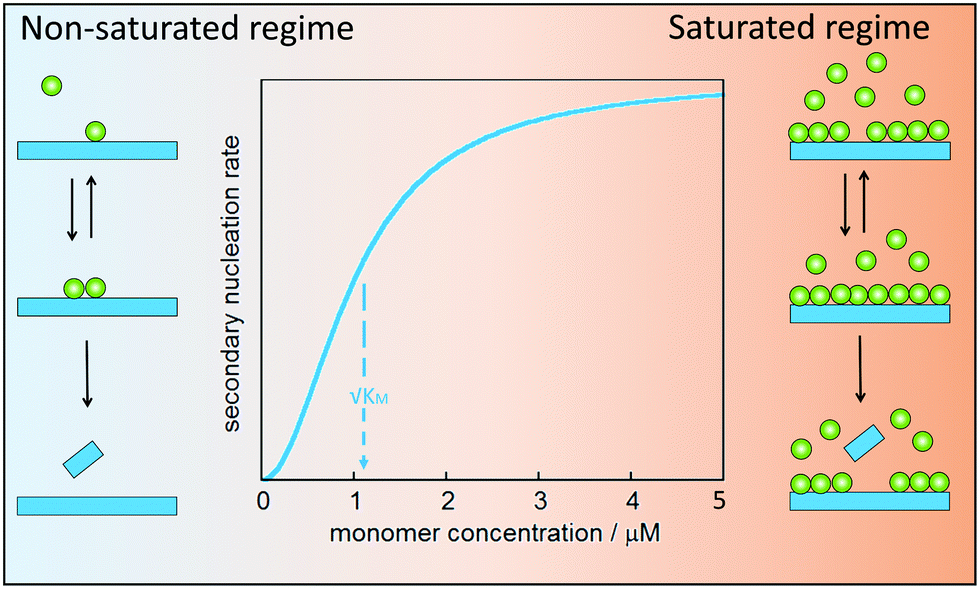 | ||
| Fig. 8 Multi-step secondary nucleation. Left: At low monomer concentration, the process is unsaturated and the observed overall aggregation profiles are strongly dependent on monomer concentration. Right: At high monomer concentration, the process is saturated and the observed overall aggregation profiles show little dependence on monomer concentration. | ||
Depending on the ratio between monomer concentration and available surface area (number of sites for secondary nucleation), any one of the rearrangement or detachment steps may become rate-limiting at high monomer concentration. Multi-step secondary nucleation share many features with Michaelis–Menten kinetics of enzyme activity such as a strong rate dependence on monomer concentration in the unsaturated regime at low monomer concentration, while in the saturated regime at high monomer concentration the rate of secondary nucleation is independent of monomer concentration.79 The kinetic modelling of saturated secondary nucleation includes the equivalent of a Michaelis constant, KM, which indicates the monomer concentration at which the process is half saturated (Fig. 8).79 Saturation of secondary nucleation (multi-step secondary nucleation) leads to a change in scaling exponent in plots of half-time (the time it takes until half he monomer has converted to fibrils) versus initial monomer concentration, a feature which can not be explained by models including only primary nucleation and singe-step secondary nucleation.79,93,95,98
Most likely the process is more complex than pictured in Fig. 8. For example, the surface of the aggregate may serve as an oligomer generator with the final transformation/nucleation to an aggregate with the same structure and growth rate as the fibril occurring after detachment. Alternatively, monomers may bind to the surface where small clusters form and nucleate/transform to fibril structure before they detach and grow.99 The nucleated species may also continue to grow along the aggregate surface before they detach. In a fourth scenario, clusters of monomer could form in solution and nucleate when they come in contact with the aggregate surface.64,79,99
3.6 Oligomer definitions
The definitions for oligomers vary in the literature from being defined by size (e.g. 2 to 20-mers or other size range), growth rate (lower growth rate than fibrils), structure (less organized structure than fibrils) or biological activity (more toxic than fibrils). Here we discuss oligomers that have less ordered structure and a lower growth rate than fibrils, and we will focus on oligomers that form during an ongoing amyloid formation process, in contrast to oligomers defined according to preparation methods using substances that block them from further growth.3.7 Oligomer generation & toxicity
A critical feature of secondary nucleation is that it can generate significant concentrations of oligomeric aggregates. This is because the rate of secondary nucleation depends on the amount of fibrils formed, which in turn grows exponentially with time, at least during the early times of aggregation (see Fig. 9 and associated discussion). It is thus of critical importance to understand the mechanisms of oligomer generation by secondary nucleation. Secondary nucleation is likely to generate oligomeric aggregates with a wide range of sizes and structures. The products of secondary nucleation are thus aggregates that are smaller than the mature fibrils, with less organized structure and lower growth rate. For example, recent single-molecule studies of oligomers of Ure2p generated by primary nucleation have highlighted that these oligomeric populations consist of relatively disordered oligomers that either dissociate back to monomers or undergo a structural conversion into more structured oligomers, which, in turn, can convert to elongation-competent fibrillar structures. A common feature of oligomers generated by primary and secondary nucleation seems to be that they initially take the form of less structured aggregates that then undergo a conversion into growth-competent species. In the case of primary nucleation, this mechanism has been observed in simulations of coarse-grained model systems,38–40 in atomistic simulations of tau100 and Aβ101 fragments, and experimentally in the case of the yeast prion protein Ure2.102 Simulations of coarse grained systems have also revealed that secondary nucleation may involve the structural conversion of disordered oligomers to ordered aggregates.64,99 The existence of a conversion step of oligomeric species into growth-competent fibrillar species is supported by experiments, in which case measurements of oligomer populations, generated by secondary nucleation during Aβ42 aggregation were coupled to kinetic analysis,103 see also Section 4.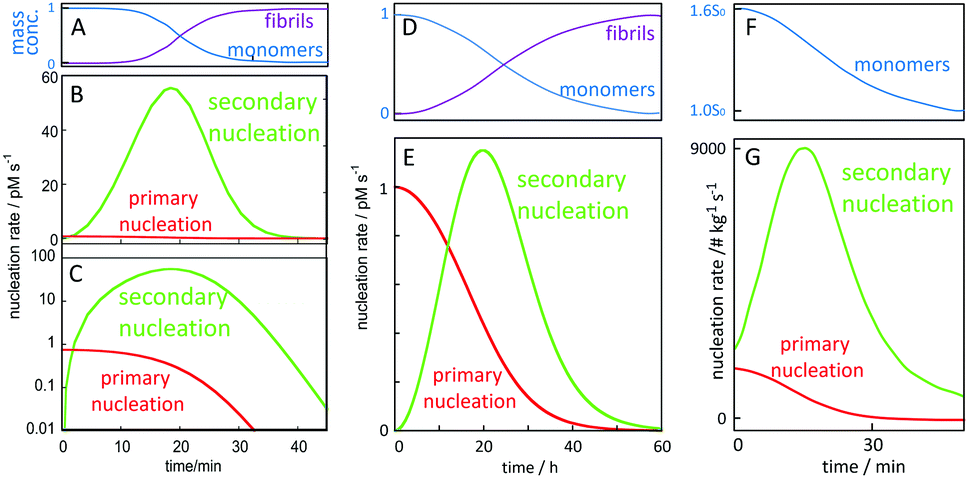 | ||
| Fig. 9 Time-dependence of the macroscopically observable monomer and fibril concentration as a function of time and (B, C and E) nucleation rates. Starting from 5 μM (A–C) or 100 nM (D and E) Aβ42 monomer in 20 mM NaP, 150 mM NaCl, pH 8.0 for, the rate of primary nucleation (red), depends on monomer concentration only, and has its maximum at time zero and remains relatively constant over the lag phase where the monomer concentration (A and D blue) remains almost intact. Secondary nucleation (green) requires both aggregate and monomer and has its maximum rate close to mid-point of the overall aggregation curve. (F and G) Starting from 1.6 times supersaturated paracetamol with seeds added at time zero, the rate of secondary nucleation (green) is always higher than that of primary nucleation (red). Curves in panels A, B, C, F and G are adapted from ref. 112 and 113. | ||
Many studies have indicated that toxicity is associated with protein oligomers or the ongoing process leading to their generation.104–107 Most intriguingly, toxicity seems to arise most prominently during the aggregation process in a reaction involving both fibrils and monomers.78,108,109 Electrophysiology measurements of γ oscillations in rat brain slices, a process involved in memory and learning, revealed a strong toxic effect from oligomeric Aβ42 species produced due to monomer-dependent secondary nucleation.109 While amyloid fibrils do not seem to be toxic per se, fibrils might still be considered disease-relevant species,110 possibly by presenting an oligomer-breeding surface.
In order to gain quantitative insights into the rate of oligomer generation during an ongoing aggregation reaction, it is useful to calculate the nucleation rate in a given experimental setup. In Fig. 9, we have calculated the rates of primary and secondary nucleation as a function of time for in vitro reactions starting at time zero from a defined monomer concentration using the values for the rate constants for primary nucleation, secondary nucleation and elongation, as obtained from global fitting to aggregation kinetics data for Aβ42 over a range of different peptide concentrations.78,111 These calculations reveal that the rate of primary nucleation is maximal at time zero, when only monomer is present, and, in the absence of preformed fibrils, dominates over secondary nucleation during the very early stages of aggregation, when the aggregate concentration is low. By contrast, the rate of secondary nucleation, which depends on the aggregate concentration, is zero initially and overtakes primary nucleation only after a critical concentration of fibrils is reached. Dimensionality arguments show that this critical aggregate concentration above which secondary nucleation dominates the production of new fibrils is equal to the ratio of the primary and secondary nucleation rates. Eventually, over the time course of the reaction, secondary nucleation may therefore generate many orders of magnitude more oligomers than primary nucleation. Because the rate of secondary nucleation (Fig. 9B, C and E) depends on both aggregate and monomer concentration, it has its maximum close to the mid-point of the growth phase of the macroscopic aggregation curve, where both monomer and fibril concentration are close to 50% of the total monomer concentration (Fig. 9A and D). Very similar curves can be presented for the rates of primary and secondary nucleation in the presence of preformed aggregates, such as in the case of paracetamol (Fig. 9F and G).112 In this case, if a sufficient amount of seed is added initially, the rate of secondary nucleation may dominate the production of new aggregates over the entire duration of the reaction.
3.8 Intra-cellular targets of toxic oligomers that form during an aggregation reaction
The calculated nucleation rates (Fig. 9), can be used to guide the experimental setup for finding oligomer-specific interaction partners. Such design, aiming to cover the time-frame during which the majority of toxic oligomers are generated, was recently used in protein array screening for intracellular targets of Aβ42 oligomer generated by secondary nucleation during an on-going aggregation reaction.113 Using the rate constants, determined in physiological salt buffer at 37 °C,93 it was calculated that most of the toxic oligomers would be generated between 8 and 23 minutes of a reaction starting from 5 μM monomer at time zero (Fig. 10A). An upside-down orientation, with the array placed on top of the reaction solution, was a critical feature of the experimental setup to avoid fibril sedimentation onto the array. This approach lead to the identification of one target significantly above the noise – glycogen synthase kinase 3α (GSK3α, Fig. 10B and C). An interaction between Aβ42 oligomers and GSK3α, and between Aβ42 oligomers and the close homologue GSK3β, was validated using thermophoresis, surface plasmon resonance and phosphorylation assays.113 The results are intriguing in the light of the protein tau, another main player in Alzheimer's disease, being one of the substrates that gets phosphorylated by GSK3α and GSK3β.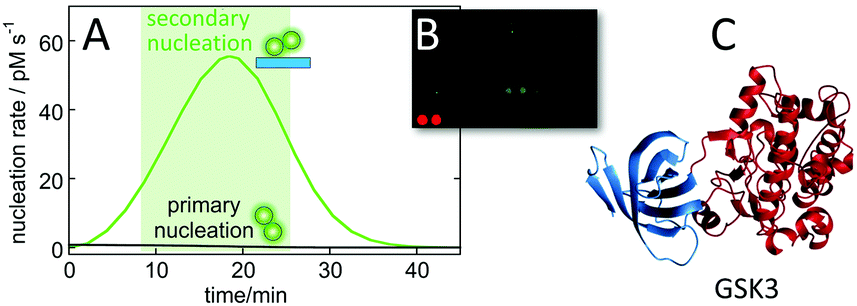 | ||
| Fig. 10 Identification of GSK3α as an intracellular target of Aβ oligomers. Protein arrays were incubated between 8 and 23 minutes into an ongoing aggregation reaction of 5 μM Aβ42 (A), with a minor fraction being fluorophore labelled to facilitate imaging (B) leading to identification of one putative target significantly above back-ground (C).113 | ||
3.9 Secondary-nucleation inhibitors
The strong connection between monomer-dependent secondary nucleation and generation of toxicity, motivates the search for inhibitors that specifically suppress this microscopic step. Inhibitor failures at late stages of development have been taken as an argument against the amyloid cascade hypothesis;114,115 however, these inhibitors were identified before the Aβ aggregation mechanism was known with an aim to retard the overall macroscopically observable aggregation process. Recent advances have now made it possible to instead search for inhibitors that selectively target the nucleation steps (Fig. 11A and B), such that the generation of toxic species is either delayed (Fig. 11D) or reduced (Fig. 11E), while inhibitors of elongation (Fig. 11C), which may even increase toxicity over time (Fig. 11F), can be discarded early in the discovery process.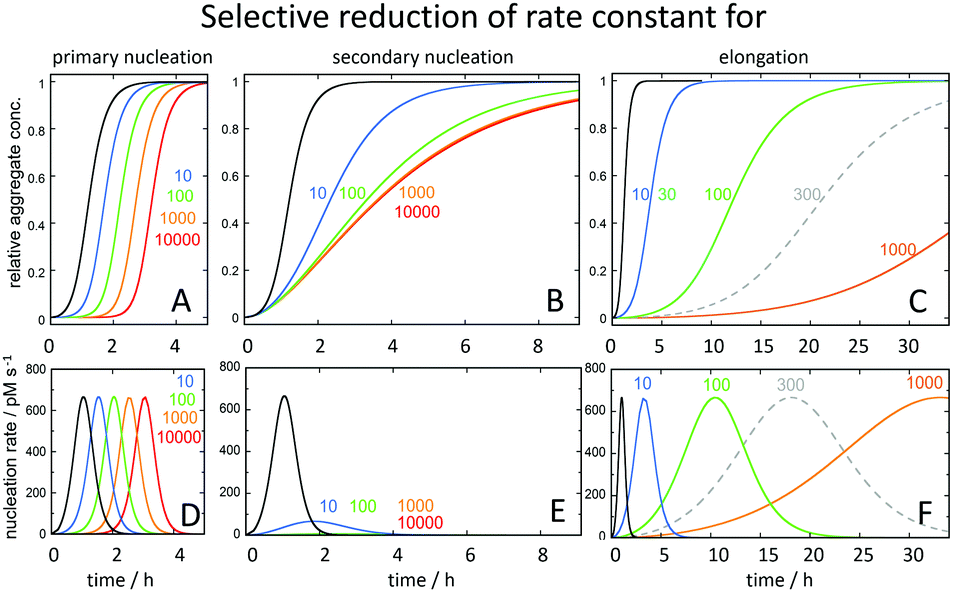 | ||
| Fig. 11 Predicted changes in the macroscopic aggregation curves upon selective reduction of the rate constants of (A) primary nucleation (kn), (B) secondary nucleation (k2) and (C) elongation (k+) calculated using the Amylofit platform.117 All calculations assumed that the reaction was initiated at time 0 from a solution containing 3 μM Aβ42 monomer. The reference black curves in each panel were calculated using the rate constants measured for Aβ42 in 20 mM sodium phosphate buffer with 0.2 mM EDTA, pH 8.0, 37 °C: kn = 3 × 10−1 M−1 s−1, k2 = 1 × 104 M−1 s−1, k+ = 3 × 106 M−1 s−1.78 For each coloured curve, two rate constants were fixed at these values and the fold-reduction of the selected microscopic rate constant colour coded as shown above the panels. In panels D, E and F is shown the total nucleation rate (primary plus secondary) as a function of time for the same selective rate constant reductions. | ||
Inhibitors may suppress a single microscopic step in the overall aggregation process, or may act on more than one step, depending on whether the inhibitor interacts with monomers, oligomer or fibrils.116 Monomer-binders may block any one process, and fibril-binding molecules may block secondary nucleation or elongation.
The screening for inhibitors that specifically suppress secondary nucleation is facilitated by the fact that very different effect on the macroscopic aggregation curves can be expected upon inhibition of this process compared to inhibition of primary nucleation or elongation (Fig. 11). Inhibition of secondary nucleation leads to a reduced slope of the growth phase and this effect saturates at high inhibitor concentrations (Fig. 11B).
The first example of a specific inhibitor of monomer-dependent secondary nucleation of Aβ42 was the molecular chaperone domain pro-SPC Brichos, which inhibits this process in a highly selective manner by binding to the fibril surfaces.109 Electrophysiology measurements show that the toxicity related to oligomers generated in an ongoing aggregation reaction is blocked in the presence of pro-SPC Brichos.109 Further work has shown that secondary nucleation inhibitors can be produced in the form of antibody fragments either through rational design towards discrete regions of the Aβ42 sequence118 or through selection from phage-display libraries using a combination negative selection to remove monomer binders and positive selection to retain fibril binders.119 The coupling to kinetic screening allowed identification of antibody fragments (scFvs) that inhibit the secondary nucleation of Aβ42 on fibril surfaces in a specific manner, while disregarding scFvs that inhibit elongation to ensure suppression rather than enhancement of oligomer generation.119 The finding of both kinds of antibodies, moreover implies that the “sites” for secondary nucleation and elongation are not the same.118,119 Inhibition of secondary nucleation of Aβ42 or Aβ40 has also been observed with small molecules.120,121 On the contrary, the cellular prion protein has been found to inhibit elongation with no effect on secondary nucleation.122 Secondary nucleation of α-syn can be inhibited by the homologous protein β-synuclein, which competes with α-syn in binding to the fibril surface.123
3.10 Insights from studies of Aβ variants
Aβ exists in body fluids like blood and cerebrospinal fluid as a range of length variants125,126 besides the main species with 40 or 42 residues. As described above, both Aβ40 and Aβ42 display efficient monomer-dependent secondary nucleation.78,79 The relative importance of secondary nucleation is higher in Aβ40 compared to Aβ42 because of dominating suppression of primary nucleation.79 Secondary nucleation has been inferred also for variants of N-terminally extended Aβ42 with up to 40 extra residues from the precursor protein127 and for N-terminally truncated Aβ5–42128 and Aβ3–42 with pyroglutamate at the N-terminus.129 These studies show that the exact length of the peptide is not critical for secondary nucleation and that secondary nucleation is retained even if the monomer and fibril are decorated with an additional equal number of residues at the N-terminus.There are several known familial mutations, which lead to increased risk of Alzheimer's or related diseases.124 One such mutation is A2V, which increases the hydrophobicity of the N-terminal part of the peptide, and has been observed to increase Aβ production with enhanced aggregation in vivo.130In vitro mechanistic studies of Aβ42 A2V show that the overall rate of aggregation is largely unaffected. However, this mutation enhances the relative importance of secondary over primary nucleation, such that the secondary process becomes even more dominant compared to Aβ42 wild-type and saturates at lower monomer concentrations than the wt.95 Other very aggressive Aβ variants contain the so-called Arctic mutation E22G and the Iowa mutation D23N, both of which diminish the electrostatic repulsion between monomers and between monomers and fibrils, therefore an increase in aggregation rate is expected. Recent work has shown, however, that the rate constant for secondary nucleation is increased more than those for primary nucleation and elongation, with saturation of secondary nucleation for Aβ42 with E22G96,97 and D23N.97 Moreover, the effect of these mutations is more severe the lower the peptide concentration and the half-time for fibril formation is shortened by more than two orders of magnitude relative to WT in measurements at peptide concentrations approaching the physiological range. Although these mutations occur very close to one another in the peptide sequence and on the fibril surface, D23N leads to a more significant loss of electrostatic repulsion than E22G.97 This finding might seem surprising in light of the long-range nature of electrostatic interactions and may indicate that the contribution to the electrostatic repulsion from the E22 side-chain is modulated by its closeness to the K16 side-chain on the fibril surface (Fig. 12). An enhanced rate of monomer-dependent secondary nucleation has also been observed for Aβ42 with the so-called Dutch and Italian mutations E22Q and E22K.97
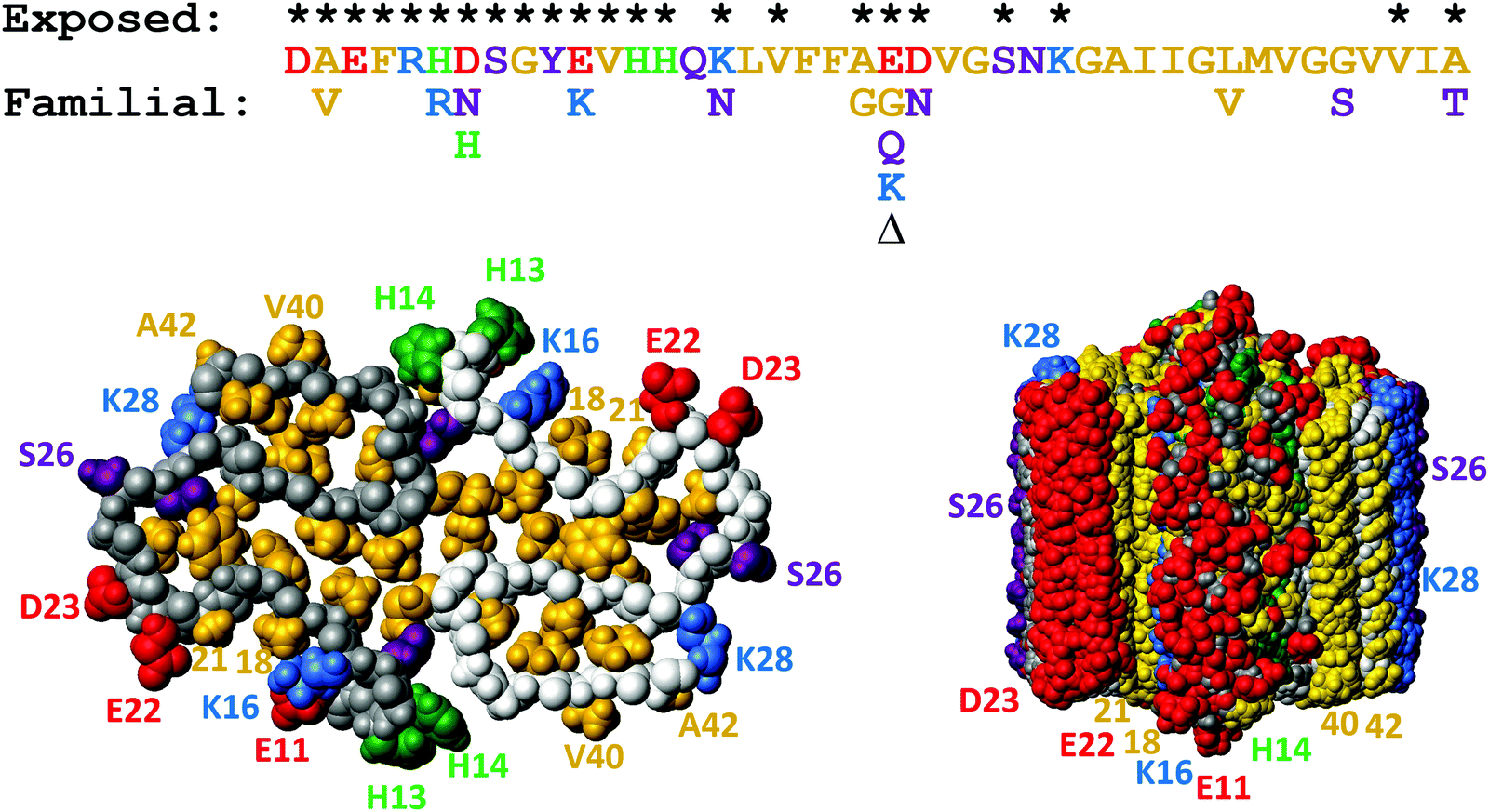 | ||
| Fig. 12 Top: Aβ42 sequence with the sites of familial point mutations associated with disease124 shown below. Bottom: Solid state NMR model of the fibrillar structure with top view of one plane with two monomers to the left and a side view of ten such planes to the right. The colors are used to indicate hydrophobic (gold), acidic (red), basic (blue), titrating (green) and hydrophilic (purple) side-chains. Residues which are exposed on the fibril surface are indicated with * above the sequence, and also numbered in the structure plots. This figure was prepared using Molmol2 based on the pdb file 5KK3.1 | ||
Two nearly identical models of the Aβ42 structure, each based on many hundreds of distance constraints, were recently presented,1,16 and serve as a source for design of inhibitory molecules as well as new mutational variants of the Aβ42 peptide. In particular relevance for understanding the molecular determinants of secondary nucleation are 9 sidechains which are exposed on the surface of the ordered part of the fibril (K16, V18, A21, E22, D23, S26, K28, V40 and A42), plus residues 1–14 in the relatively less ordered N-terminus (Fig. 12). On-going work in our laboratory addressing all these 23 residues shows that it is very difficult to design mutations that remove secondary nucleation.131,132
3.11 Insights from variations in solution conditions
Understanding how variations in solution conditions affect the aggregation behaviour is crucial in extrapolating the findings from in vitro kinetics to other systems and in particular to in vivo aggregation. The presence of solutes that bind to specific parts of the protein can significantly alter the aggregation behaviour, for example leading to an increase of the relative importance of secondary nucleation by suppression of elongation.133 More generally, the effects of ionic strength and pH are two fundamental aspects governing protein aggregation. The monomeric state of the aggregating protein is often charged and thus the aggregation involves the association of like charged states, disfavoured by electrostatic repulsion. In general, changes that lead to a decrease in the charge repulsion effect are found to speed up aggregation, in particular the processes that involve the encounter of several charged species. These changes can take the form of a change in pH95 or an increase in ionic strength.93,94 In addition to increasing the overall rate of aggregation, these changes often also affect the balance of the individual steps in multistep processes, such as secondary nucleation. As secondary nucleation involves an attachment step, followed by a rearrangement/detachment step, decrease in charge repulsion often speeds up the initial attachment step more than the subsequent steps, thus leading to a saturation of secondary nucleation.93,953.12 Specificity
As described above, both of Aβ40 and Aβ42 display significant secondary nucleation, which is observed as a very significant shortening of the lag phase upon addition of sub-1% (by mass) of pre-formed seeds. Several studies have investigated mixtures of Aβ40 and Aβ42 of varying origin and purity, with acceleration of Aβ40 aggregation in the presence of Aβ42 monomers and a slight retardation of Aβ42 aggregation in the presence of Aβ40 monomers as a convergent result (ref. 134 and references therein). A recent study using ultra-pure recombinant peptides showed that fibrils of each length variant fail to accelerate the aggregation of the other variant.134 Aβ40 monomers fail to nucleate on fibrils of Aβ42 and Aβ42 monomers fail to nucleate on fibrils of Aβ40, thus there is no cross-catalysis of nucleation (Fig. 13A). | ||
| Fig. 13 Cross- versus self-seeding data for (A) Aβ40 vs. Aβ42 and (B) Aβ42 vs. 30Aβ42 – a 30 residue N-terminally extended variant of Aβ42. | ||
In contrast, an acceleration of Aβ40 was observed in samples containing also Aβ42 monomers, relative to samples with the same concentration of Aβ40 alone. Moreover, reactions starting from mixed monomers display macroscopic aggregation curves with two sigmoidal curves when monitored by ThT fluorescence. Mass spectrometric analysis of selectively isotope-labelled samples identified the first transitions as Aβ42 fibril formation and the second one as Aβ40 fibril formation (Fig. 14).134
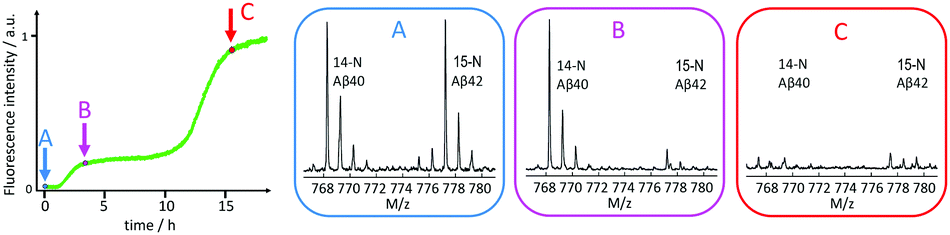 | ||
| Fig. 14 Mass spectrometric quantification of the relative amount of 14N-Aβ40 and 15N-Aβ42 monomers remaining in solution after centrifugation to remove any formed fibrils at time zero (A), after the first sigmoidal transition (B), and after the second sigmoidal transition (C) as observed by ThT fluorescence. At time zero, the solution contained 1.5 μM 14N-Aβ40 monomers and 1.5 μM 15N-Aβ42 monomers. Clearly Aβ42 is selectively consumed during the first transition, which is thus identified as Aβ42 fibril formation, and the second transition is identified as Aβ40 fibril formation.134 | ||
The acceleration of Aβ40 aggregation in the presence of Aβ42 monomers, but not Aβ42 fibrils, implies that the two peptides cross-react at the stage of primary nucleation only. This remarkable result implies that monomer-dependent nucleation on fibrils is not a general surface effect but dependent on the detailed structure of the aggregate. While two independent models of Aβ42 at physiological pH in water-based buffers are nearly identical,1,16 and also compatible with a much smaller number of distance constraints in an earlier investigation,135 there are multiple models of Aβ40 in the literature.12,17,136–139 Still, the tight packing of the C-terminus and thereby fibril core of Aβ42, with high structural order of all amino acid up to the very end suggest that there would be very dramatic destabilization of this fold for the Aβ40 peptide lacking both Ile41 and Ala42. Opposite to this, many of the Aβ40 models display the C-terminus in a conformation where steric clashes prohibit the incorporation of two extra residues.
The lack of cross-seeding between Aβ40 fibrils and Aβ42 monomers, and vice versa, was confirmed in a recent study, which also showed the emergence of cross-seeding between Aβ42 fibrils and Aβ40 monomers if Aβ40 contains the arctic mutation E22G.140 Intriguingly the fibrils formed from Aβ40-E22G monomers in the presence of Aβ42 fibrils showed the same solid state NMR spectrum as the parent Aβ42 fibrils.140
In contrast to the failure of Aβ40 and Aβ42 to form joint fibrils, variation of N-terminal length seems less detrimental. A series of N-terminally extended Aβ42 peptides cross-seed with normal Aβ42 (Fig. 13B), implying that decoration of fibrils with extended N-termini does not block the catalytic reaction, nor does N-terminal extensions of the monomer hinder its nucleation on the surface of fibrils formed from non-extended Aβ42. The extended N-termini are likely to protrude from the structured region like an unstructured polymer brush, meaning that the fibril core structure might be the same as for Aβ42.
3.13 Secondary nucleation in vivo
While much of the mechanistic work leading to the identification and characterization of monomer-dependent secondary nucleation has been performed in vitro with highly pure and homogeneous peptides and proteins in aqueous buffers, a key question is its relevance and importance in vivo, for example as regards spreading of pathology and toxicity to neuronal cells. The starting concentrations of monomer are also typically order of magnitude higher in in vitro experiments compared to physiological levels, while in an advanced disease state the local concentrations of fibrillar material may be very high. In vivo, monomers and aggregates exist in an environment that contains high and low concentrations of many thousands of other proteins and biomolecules, lipid membranes, electrolytes, metabolites, etc. Each one of these may affect the aggregation process in a different manner. Moreover, several amyloid proteins, including Aβ peptide, exists in vivo as a collection of length variants. However, most likely the same underlying microscopic steps exist also in the aggregation reaction in a complex situation, although the rate of each step will be governed by the net effect of many different factors. Moreover, the species concentrations and reaction rates will likely change over time much more slowly since the concentrations of many proteins have a relatively slow variation as regulated by catalytic and catabolic events.There are thus all reasons to believe that secondary nucleation of monomers on the surface of fibrils occur also in vivo in body fluids and tissues. In support of this, a recent investigation using a range of optical probes found that most Alzheimer patient brains include aggregates of a single morphology, or of two discrete morphologies in physically separated locations.141 The variability of the aggregate forms was also found to be much lower within each patient than between different patients. Recent studies of Aβ42 aggregation in cerebrospinal fluid (CSF) and in the presence of phospholipid membranes, indicate that secondary nucleation is the dominating nucleation process for Aβ42 also in these conditions.142–144
In vivo, aggregation occurs in the presence of phospholipid membranes enclosing cells, organelles and extracellular vesicles. In the case of α-syn, this has been established as a key environment promoting heterogeneous primary nucleation, as recently reviewed.145 Much of the early work on the aggregation of Aβ peptides in the presence of phospholipid membranes employed synthetic peptide and the use of co-solvents, which may alter the membrane integrity as well as the partitioning of the peptide between solution and membrane. However, recent work using recombinant peptide avoid such artefacts and have revealed that membranes may retard or catalyse nucleation of Aβ42 aggregation depending on the lipid-to-protein ratio and the membrane composition.143,144,146 The cholesterol fraction has been identified as a key factor promoting nucleation.144,146
Several lines of evidence support seeding in vivo;147–153 while this may at least in part be due to secondary nucleation, these studies have not asked this question. After noting that fibril deposits in familial Alzheimer's disease (fAD) have different conformations than in sporadic Alzheimer's disease (sAD), fAD Aβ40 fibrils (E22G or E22Q) were injected into transgenic mouse brain that carried only wild type (WT) Aβ40 gene. The plaque formation of Aβ40 WT was induced and the Aβ aggregate strain (E22G or E22Q) was inherited and finally lead to a fAD-like pathology. This indicates that the injected fibrils served as a propagation template in the spreading of Aβ pathology. Several other studies in rodent models confirm that an AD pathology can be accelerated in a dose dependent manner by injecting Aβ deposit containing brain extract.147–151
An animal model closer to human was employed in a long term study, where brain homogenate from human or marmoset with sAD was injected into marmoset brain.151 The experiment was followed for up to 10 years, and the vast majority of injected marmosets have developed cerebral amyloidosis, while in the control group a minority had developed AD pathology.
While Aβ brain deposit has been repeatedly found to catalyse the amyloidosis in various animal models, the same phenomenon was accidentally studied in humans. Before 1985, human cadaveric pituitary-derived growth hormone (hGH) was commonly used to treat young individuals with growth hormone deficiency. Hundreds of recipients caught Creutzfeldt–Jakob disease (CJD) due to the contamination of the hGH sample. In a post-mortem study of iatrogenic CJD patient, Aβ deposit was surprisingly observed in the brain tissue, which indicates that the hGH sample extracted from pituitary gland was not only contaminated by CJD prion protein but also Aβ aggregates.152,153
4. Insights from simulations
Simulations can provide valuable insights into monomer-dependent secondary nucleation by testing its requirements in terms of molecular nature and interaction potentials and may elucidate possible molecular events and driving forces. A number of Monte Carlo or molecular dynamics simulations have used various levels of coarse graining38–40,154–158 or atomistic representation,100,101,159 as recently reviewed,160 and provide insights into the energy barriers and transitions during primary nucleation. However, large system size and simulation time are required to capture both fibril formation and the subsequent secondary nucleation step. Simulations that address secondary nucleation therefore typically use a stating configuration that includes both preformed seeds and monomers.64,99,159,161,162 There seems to be little restrictions in terms of monomer structure since even Lennard-Jones particles, i.e. spheres, were found to undergo secondary nucleation on the surface of already formed aggregates of the same kind of particles.64 The relative strength of solute–solute, solute–solvent and solvent–solvent interactions, were varied as a means to control the level of supersaturation of the system at time zero. Interestingly, in a regime of intermediate supersaturation was found the formation of disordered monomer clusters in solution, which nucleated in contact with the surface of the parent aggregate, thereby taking up the crystal packing arrangement of the parent aggregate.64 A simulation of amyloid formation, used a more elaborate coarse-grained model in which monomers were allowed to switch between two different conformations, one of which represents the conformation in fibrils and one representing the predominant conformation in solution.99 These simulations highlighted that the nucleation of amyloid fibrils follow many but not all aspects of classical nucleation theory, for example involves at least an additional slow step corresponding to a conformational conversion of the nucleating aggregates.99 In this scenario, the nucleus size corresponds to the number of interacting monomers in the aggregate necessary to stabilize the conformational conversion step.99 In correspondence with the simpler system,64 it was found that a key determinant for secondary nucleation is the affinity of monomers for the surfaces of fibrils, and efficient self-replication takes place only in a very narrow regime of inter-monomer interaction strengths.99The results of molecular dynamics simulations of six Aβ42 monomers in the presence of a fibril fragment composed of twenty Aβ17–42 peptides (2BEG.pdb) imply that the hydrophobic fibril region causes the structural changes required for catalyzing the formation of β-sheet-rich Aβ1–42 oligomers on the fibril surface.161 These results thus reveal one plausible molecular basis of the secondary nucleation pathway. A recent all-atom molecular dynamics simulation of secondary nucleation of Aβ9–40 studied the association of monomers, dimers, etc. to the surface along the sides of fibrils,159 represented by the ordered part (residues 9–40) of an early solid state NMR model of Aβ1–40 fibrils with two monomers per plane (2LMN.pdb). This simulation found the hydrophobic effect (entropic gain upon release of hydration water molecules) as a major driving force for association of monomers etc. with the fibril sides.159
5. Kinetic experiments and analysis
5.1 Kinetic experiments
The discovery of secondary nucleation, as well as studies aimed at understanding molecular determinants and driving forces, require reproducible kinetic assays and well-defined starting states. The requirements go hand in hand and can luckily be optimized in parallel, as both reproducibility and defined starting state can be achieved using ultra-pure and sequence-homogeneous monomer preparations.163 Recombinant expression offers superior sequence homogeneity over peptide synthesis, relying on the much superior fidelity of the protein translation machinery relative to synthetic coupling steps. Expression of the aggregation prone Aβ is facilitated by the formation of inclusion bodies, which protects against degradation of the peptide and allows large quantities to accumulate in E. coli cells without significant inference with their growth. High purity Aβ can be achieved using repeated sonication, inclusion body solubilization in urea, ion exchange (IEX) in batch mode and size exclusion steps, essentially as described,164 using IEX elution with 50–75 mM NaCl and repeated gel filtration steps.78 The peptide is considered pure enough when no other proteins can be detected by silver-stained electrophoresis gels, no other molecules can be detected by 1H NMR and only peptide with the correct Mw for full-length peptide can be observed by mass spectrometry.164 Pure monomer is then isolated from such samples using gel filtration just prior to starting the kinetic experiment. High reproducibility of the latter can be achieved through extensive degassing of the buffer used, inertness of surface material used (e.g. PEGylated rather than plain polystyrene), minimization of all surface areas, including that of the air–water interface, use of highest grade ThT, optimization of the ThT concentration to be in a regime where the fluorescence intensity is proportional the mass concentration of fibrils formed.78,163 Moreover, it is essential to record aggregation data both as a function of time and at several initial concentrations, with better coverage if the latter varied such that each subsequent pair of concentrations are related by the same multiplication factor, rather than a linear variation.5.2 Kinetic models and data analysis
At the basis of connecting experimental data with mechanistic models of aggregation lies a mathematical description of the kinetics of the phenomenon. The IUPAC definition of the term reaction mechanism states “An acceptable mechanism […] must be consistent with […] the rate law”.165 Thus, the development of integrated rate laws is crucial in obtaining a mechanistic model of aggregation. Different models are derived starting from a master equation that describes the time evolution of the concentration of all species in the reaction network of aggregation.83 This description can be simplified considerably by reducing it to the experimentally observable quantities, the total aggregate mass and aggregate number. Solutions to the resulting differential rate equations for these two quantities can then be derived and used to fit the data.83–86 Due to the complexity of the models of aggregation, this data analysis should be performed in a global manner, such that the equations fit the kinetics at several monomer and seed concentrations simultaneously. A software that automates many of these tasks is available online.117 Another aspect of a successful determination of mechanism is the acquisition of data of sufficiently high quality to enable a robust analysis. Important aspects include: (1) sample purity to ensure the absence of any other substances, oxidized or degraded proteins and, most crucially preformed aggregates. (2) Optimization of the reporter dye, to verify that there is a linear correlation between signal and fibril mass and that the presence of the reporter dye does not affect the aggregation kinetics. (3) Control of conditions at all stages of preparation and during the experiment to avoid the introduction of reactive surfaces, such as air bubbles, or the initiation of aggregation during sample preparation.6. Outlook – outstanding questions regarding secondary nucleation in amyloid systems
While secondary nucleation has been known for a long time in non-amyloid systems its discovery in amyloid aggregation is still relatively recent. For this reason, a number of key questions pertaining uniquely to secondary nucleation in these low-dimensional systems remain more or less open. We will here take the opportunity to discuss a few of these questions, and to propose some possible answers. We will also outline some potential future directions for research in this field.6.1 Does secondary nucleation happen along the fibril surface?
Elongation occurs at the end of the fibrils; however, the catalytic surface for secondary nucleation may instead be found along the sides of the fibrils (Fig. 15A). A first support of this hypothesis are the results for the chaperone domain Brichos, which is a potent inhibitor of secondary nucleation and by immuno-gold TEM, Brichos appears to bind along the sides of fibrils.109 As a second line of support, several antibodies that selectively inhibit either elongation or secondary nucleation, have been identified after rational design as well as from random screens, which implies that these two processes do not occur at the same sites.118,119 There are also results from high resolution microscopy, including dSTORM,81 AFM166 and cryo-EM167 that seem to show transient binding events on the sides of the fibrils.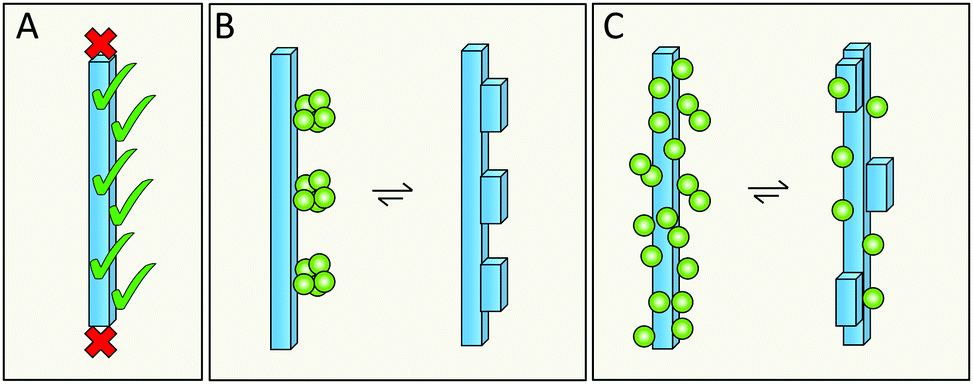 | ||
| Fig. 15 The catalytic surface for secondary nucleation is present along the sides of fibrils (A). This may take the form of distinct sites (B), or be a more diffuse aspect of the surface (C). | ||
6.2 Does secondary nucleation occur at defined sites or is it a diffuse process?
Specific catalytic sites for secondary nucleation may exist at random defects in the fibril structure or at well-defined locations defined by the molecular structure of the surface and the fibril architecture (Fig. 15B). In support of this hypothesis is the high degree of specificity that has been observed in secondary nucleation.134 On the other hand, the periodicity of such specific sites may be as small as the repeat distance of the fibril, i.e. one plane of 4.5 Å. This would mean that even if the sites are well-defined, they will be grossly overlapping with each other as each site is likely to include more than one plane of the fibril. There is also the opposite possibility that there are no specific sites, in which case incoming monomers associate diffusely along the fibril surface (Fig. 15C) and secondary nucleation would be mainly an effect of the increased local concentration at the fibril surface. This latter possibility is, however, more difficult to reconcile with the structural specificity of the process.1346.3 Does secondary nucleation involve the highly ordered fibril core structure or the flexible termini that decorate the fibril?
Related, but not identical to the previous question is whether specific residues or features that are exposed on the fibrils surface take part in secondary nucleation. Building on the enzyme analogy, secondary nucleation may require precise positioning of key residues in space for efficient catalysis. This would then suggest the involvement of residues with well-defined structural positions on the fibril surface (Fig. 16A). In several amyloid fibrils, including those of Aβ and α-syn, these exposed residues are of two kinds – more disordered and more rigid. In the case of Aβ42, the exposed sidechains in relatively highly ordered structure are K16, V18, A21, E22, D23, S26, K28, V40 and A42 (Fig. 12).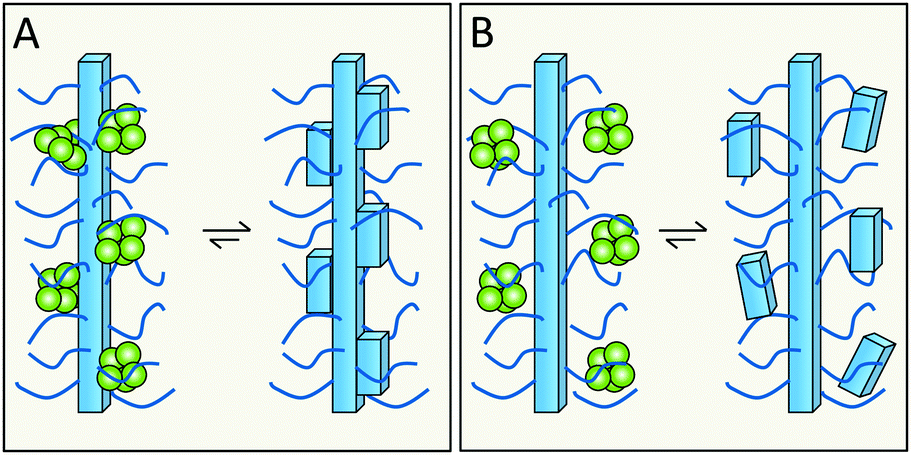 | ||
| Fig. 16 Does monomer-dependent secondary nucleation involve the more ordered (A), or more flexible (B), parts of the surface? | ||
The relatively less ordered N- or C-termini of amyloid protein monomers in fibrils form a flexible polymer brush decorating the fibrils. In case of Aβ42 this includes residues D1–H14, and for α-syn a very long C-terminus of ca. 40 residues plus a shorter N-terminus. These polymer brushes may create an environment in which incoming monomers are enriched and nucleate (Fig. 16B). In the case of enzymes, mutational studies have been of great importance in the identification of key catalytic residues. Analogous studies are ongoing for various amyloids but have this far failed to remove monomer-dependent secondary nucleation, implying that this either is a very fundamental property or an emergent property of the system.
6.4 Do oligomers form in solution or at the fibril surface?
Simulations in a minimal system using Lennard-Jones particles64 have revealed a mechanism by which relatively large disordered clusters form in solution at intermediate levels of supersaturation. Upon encountering the surface of a seed crystal a disordered cluster may undergo crystallization and take on the structure of the seed. One might envision a similar mechanism for secondary nucleation in amyloid aggregation, in which disordered oligomers form in solution and undergo structural conversion upon encountering the surface of a mature fibril (Fig. 17A, kinetic description in ref. 79). Alternatively, peptide monomers may associate with the fibril surface and oligomerization might occur on the fibril (Fig. 17B and C, kinetic description in ref. 99), with a rate dependent on the amount of monomers bound per surface area.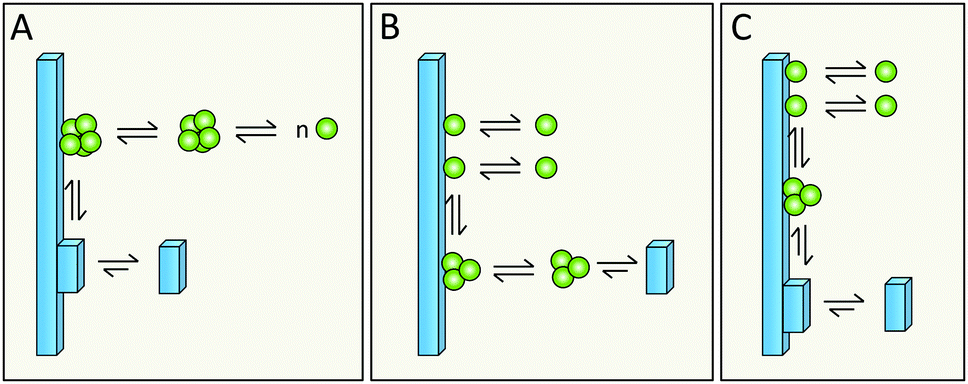 | ||
| Fig. 17 Does the structural conversion to fibrillar form happen on the surface (A and C) or in solution (C) and does the protein arrive to the surface in the form of monomer (B and C) or in the form of monomer clusters (A)? | ||
6.5 Does structural conversion happen before or after detachment?
It is possible that the step catalyzed by the fibril surface is the association of monomers into oligomeric clusters (Fig. 17B and C). Once formed these clusters may either undergo conversion into the fibrillar structure in solution after detachment (Fig. 17B) or while attached to the parent fibril (Fig. 17C). The second scenario is highly plausible; formation of new fibrils that adopt the precise structure of the parent fibril is most favored, since the interfacial energy, the molar volume of the solid phase and the difference in chemical potential between the solution and solid phase will be most favorable if the new aggregate takes on the same structure as the parent;71,72 this includes adopting the same monomer fold and inter-monomer arrangement as in the parent aggregate. The second scenario (Fig. 17C) is in keeping with the well-established phenomenon of strain propagation. In the first scenario (Fig. 17B) the detaching species will be in a pre-fibrillar oligomeric form, which is widely suspected to be the main toxic species in amyloidosis.104–107 This would then be in keeping with the observed increase in toxicity of mixtures of fibrils and monomers over pure preparations of either.78,109 Pre-fibrillar oligomers may be in exchange between solution and fibril surface and convert to fibrillar form on the fibril surface and then detach as in Fig. 17A. It is of course possible that all these options occur in parallel and that their relative proportions may vary between different amyloid protein and will also depend on the solution conditions.6.6 What is the driving force for detachment?
One of the structural hallmarks of mature amyloid fibrils is their unbranched nature. This implies that even if the catalytic formation of new fibrils happens at high rates along the sides of parent fibrils, it is always followed by efficient detachment. Local packing interactions, which are essentially identical between all monomers in the aggregate, dictate the morphology at larger scale such as for example twist distance, twist chirality, the number of monomers per filament plane, and the number of filaments winding around each other in a fibril. Although some variation in the latter parameter is observed,168 there seems to be an upper limit to the number of filaments in a fibril for every amyloid system.One plausible scenario is that the nucleating species grows as a new filament, winding around the parent fibril. This new filament must have the same monomer–monomer arrangement per plane as the template to be able to build an additional layer; however, it will detach above a defined size (Fig. 18A) in the limit where additional planes would suffer from stain and steric constraints; this will eventually force the new fibril to detach. If this hypothesis is true, the efficiency of nucleation of D-peptides on L-peptide fibrils, and of nucleation of L-peptides on D-peptide fibrils, would be much reduced compared to the two homo-chiral secondary nucleation situations.
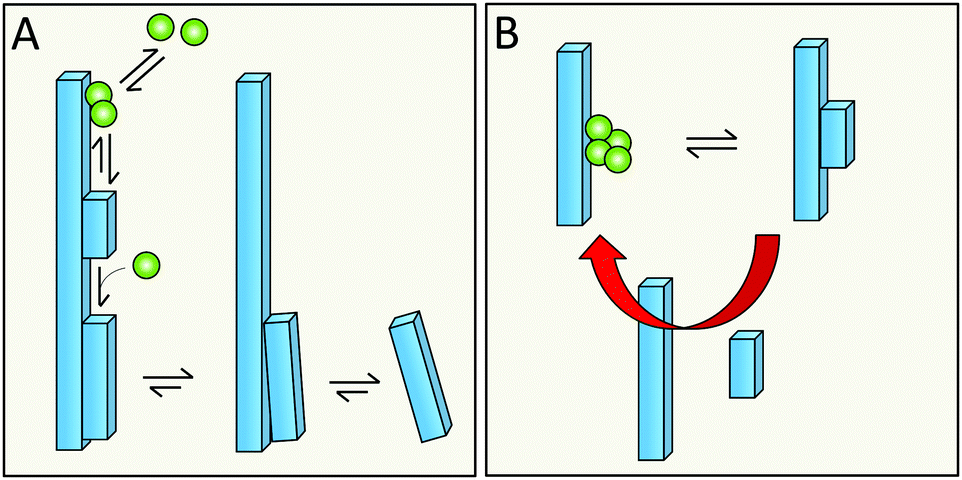 | ||
| Fig. 18 Are steric clashes a driving force for detachment (A)? Is detachment sped up by mechanical agitation (B) and thereby increasing the rate of secondary nucleation by revealing more catalytic surface at higher rate? | ||
It is also plausible that the rate of detachment is sped up by mechanical agitation (Fig. 18B), which used to be common practice in aggregation studies and is often used in industrial crystallizers. Even under quiescent conditions, which are preferred in mechanistic studies, there will be some convection in the solution. The effects of mechanical perturbations have been discussed in several studies of non-amyloid systems; the rate of secondary nucleation may increase upon increased shear flow or upon contact with foreign objects that serve to remove the newly nucleated species from the crystal surface,53,61,65 thereby making room for new catalytic events. In the case of amyloid growth, a challenge for the future will be to disentangle the expected increase in secondary nucleation rate from an increase in fragmentation rate.
6.7 Is secondary nucleation the origin of strain propagation?
Strain propagation is a well-known phenomenon in both amyloid and non-amyloid systems, i.e. the new aggregates that form in the presence of old aggregates copy the 3D structure of the old aggregate. It is well-known that seeding of a supersaturated monomer solution with pre-formed crystals leads to the generation of new crystals with the same morphology, chirality, crystal packing and space group as the seed due to secondary nucleation of monomers on the original seed.54 When nucleation happens on the surface of an already existing aggregate of the same substance, a seed, the newly formed aggregate will have lowest interfacial tension if it copies the packing/morphology of the seed. Detachment and growth by monomer addition then serves to extend the new small aggregates into new seeds of the same morphology as the original seed. This also means that the aggregate structure with the highest rate of secondary nucleation, is the one that is most efficiently propagated; this is taken advantage of in industrial and analytical crystallization procedures to obtain a more homogeneous product.49,61 In such reactions, old crystals, seeds, are introduced into a reactor or drop of supersaturated monomer solution. Secondary nucleation followed by detachment and growth thereby amplifies the seed into multiple crystals with identical packing.Does secondary nucleation also lead to proliferation of amyloid aggregates of a defined morphology, i.e. is the so-called strain phenomenon of amyloid also rooted in secondary nucleation as depicted in Fig. 19A? In the prion and amyloid field, this is often discussed as being due to fragmentation and growth of the broken seeds; this view may at least in part have originated from the fact that many experiments have been performed under vigorous mechanical agitation, conditions under which the generation of new aggregates is in fact dominated by fragmentation.83 However, in 1996 Leslie Orgel proposed that prion propagation may originate from monomer-dependent secondary nucleation.169
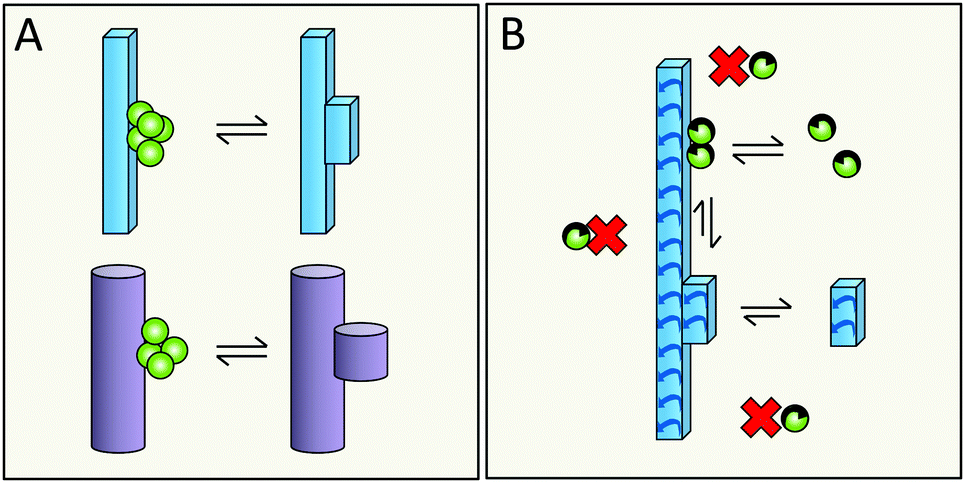 | ||
| Fig. 19 Does secondary nucleation propagate aggregate structure (A)? Is secondary nucleation enantioselective (B)? | ||
Intriguingly, a recent investigation using a range of optical probes found that most Alzheimer patient brains include aggregates of a single morphology, or of two discrete morphologies in physically separated locations.141 It would be very surprising if the strain phenomenon in amyloid formation and prion propagation is not, at least in some systems, a consequence of monomer-dependent secondary nucleation. In that case, monomer-dependent secondary nucleation of amyloid proteins and peptides might be expected to be enantio-selective.
The connection between secondary nucleation and strain propagation also ties into the discussion about structural conversion in 6.4. It implies a high degree of structural specificity which for example means that is should be enantioselective (Fig. 19B and 4), as reported in other systems.54 Several studied have demonstrated a strong enantioselectivity in amyloid seeding experiments in various amyloid systems.170–172 While these findings have mainly been discussed in terms of enantioselective elongation, it seems likely that the seed fibrils in these cases do not serve as catalysts for nucleation of the mirror peptide, since elongation of the new nuclei should then have been observable.
6.8 What is the role of secondary nucleation in amyloid pathology and spreading?
The need to understand the processes underlying spreading and toxicity in amyloid diseases is the main motivation behind the study of aggregation mechanisms. For several years the consensus in the field has been that the main toxic species in amyloid aggregation are prefibrillar oligomers. Under typical in vitro conditions oligomers generated from secondary nucleation outnumbers those created from primary nucleation by several orders of magnitude and ongoing work suggests that this is also the case in the presence of biological fluids.142 Additionally, mixtures of fibrils and monomers have proven to be far more cytotoxic than either component alone.78,108,109 A different hallmark of various amyloid diseases is the distinct pattern of spreading through adjacent tissues. The fibrils themselves are possibly not mobile enough to explain spreading, but fibril-derived secondary nuclei or small oligomeric products thereof could very well fill that role. There is hope for the future that methods to inhibit secondary nucleation could prove to be useful in mitigating both toxicity and spreading.7. Conclusions
Monomer-dependent secondary nucleation has emerged as a key step in the aggregation mechanism of many amyloid-forming peptides and proteins. The rate of this process seems to be modulated by hydrophobic and electrostatic interactions involving the amyloid proteins and their surroundings. Secondary nucleation has many similarities with enzyme catalysis as well as general surface catalysis and seems to have a high structural specificity. Its temperature dependence and thermodynamic signature is very different from primary nucleation and elongation. Moreover, secondary nucleation and elongation occur at distinct “sites” of a fibril; while secondary nucleation seems most prominent along the sides of fibrils, elongation occurs at their ends. It is therefore possible to derive inhibitors that selectively suppress secondary nucleation with little effect on elongation or primary nucleation. As the products of secondary nucleation appear to be toxic to cells and organs, these inhibitors may have significant therapeutic potential. Since its discovery during the past two decades, our understanding of monomer-dependent secondary nucleation has now reached a level where it is possible to design experiments to gain insights into the molecular determinants and driving forces of this key process, to accelerate the development of future therapeutics of amyloid diseases.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Anna Carnerup, Lund University, for help with aquiring the cryo-EM image (Fig. 1) and Lennart Lindfors, Astra Zeneca, Mölndal, for stimulating discussions. Generous support from Alzhemierfonden (SL), the European Research Council (SL, TPJK), the Swedish Research Council (SL), Peterhouse Cambridge (TCTM), Sidney Sussex College Cambridge (GM) and the Swiss National Science Foundation (TCTM), is gratefully acknowledged.Notes and references
- M. T. Colvin, R. Silvers, Q. Z. Ni, T. V. Can, I. Sergeyev, M. Rosay, K. J. Donovan, B. Michael, J. Wall, S. Linse and R. G. Griffin, J. Am. Chem. Soc., 2016, 138, 9663–9674 CrossRef PubMed.
- R. Koradi, M. Billeter and K. Wüthrich, J. Mol. Graphics, 1996, 14(51–55), 29–32 Search PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef PubMed.
- D. Eisenberg and M. Jucker, Cell, 2012, 148, 1188–1203 CrossRef PubMed.
- (a) T. P. Knowles, M. Vendruscolo and C. M. Dobson, Nat. Rev. Mol. Cell Biol., 2014, 15, 384–396 CrossRef PubMed; (b) T. C. T. Michaels, A. Šarić, J. Habchi, S. Chia, G. Meisl, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Annu. Rev. Phys. Chem., 2018, 69, 11 CrossRef PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2017, 86, 27–68 CrossRef PubMed.
- P. C. Ke, M. A. Sani, F. Ding, A. Kakinen, I. Javed, F. Separovic, T. P. Davis and R. Mezzenga, Chem. Soc. Rev., 2017, 46, 6492–6531 RSC.
- A. J. Doig, M. P. Del Castillo-Frias, O. Berthoumieu, B. Tarus, J. Nasica-Labouze, F. Sterpone, P. H. Nguyen, N. M. Hooper, P. Faller and P. Derreumaux, ACS Chem. Neurosci., 2017, 8, 1435–1437 CrossRef PubMed.
- C. B. Anfinsen, Science, 1973, 181, 223–230 CrossRef PubMed.
- C. M. Dobson, Philos. Trans. R. Soc., B, 2001, 356, 133–145 CrossRef PubMed.
- W. T. Astbury, S. Dickinson and K. Bailey, Biochem. J., 1935, 29, 2351–2360 CrossRef PubMed.
- O. N. Antzutkin, J. J. Balbach, R. D. Leapman, N. W. Rizzo, J. Reed and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13045–13050 CrossRef PubMed.
- J. J. Wiltzius, S. A. Sievers, M. R. Sawaya, D. Cascio, D. Popov, C. Riekel and D. Eisenberg, Protein Sci., 2008, 17, 1467–1474 CrossRef PubMed.
- A. K. Schütz, T. Vagt, M. Huber, O. Y. Ovchinnikova, R. Cadalbert, J. Wall, P. Güntert, A. Böckmann, R. Glockshuber and B. H. Meier, Angew. Chem., Int. Ed. Engl., 2015, 54, 331–335 CrossRef PubMed.
- J. A. Rodriguez, M. I. Ivanova, M. R. Sawaya, D. Cascio, F. E. Reyes, D. Shi, S. Sangwan, E. L. Guenther, L. M. Johnson, M. Zhang, L. Jiang, M. A. Arbing, B. L. Nannenga, J. Hattne, J. Whitelegge, A. S. Brewster, M. Messerschmidt, S. Boutet, N. K. Sauter, T. Gonen and D. S. Eisenberg, Nature, 2015, 525, 486–490 CrossRef PubMed.
- M. A. Wälti, F. Ravotti, H. Arai, C. G. Glabe, J. S. Wall, A. Böckmann, P. Güntert, B. H. Meier and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4976–E4984 CrossRef PubMed.
- W. Qiang, W. M. Yau, J. X. Lu, J. Collinge and R. Tycko, Nature, 2017, 541, 217–221 CrossRef PubMed.
- A. W. P. Fitzpatrick, B. Falcon, S. He, A. G. Murzin, G. Murshudov, H. J. Garringer, R. A. Crowther, B. Ghetti, M. Goedert and S. H. W. Scheres, Nature, 2017, 547, 185–190 CrossRef PubMed.
- M. D. Tuttl, G. Comellas, A. J. Nieuwkoop, D. J. Covell, D. A. Berthold, K. D. Kloepper, J. M. Courtney, J. K. Kim, A. M. Barclay, A. Kendall, W. Wan, G. Stubbs, C. D. Schwieters, V. M. Lee, J. M. George and C. M. Rienstra, Nat. Struct. Mol. Biol., 2016, 23, 409–415 CrossRef PubMed.
- L. Gremer, D. Schölzel, C. Schenk, E. Reinartz, J. Labahn, R. B. G. Ravelli, M. Tusche, C. Lopez-Iglesias, W. Hoyer, H. Heise, D. Willbold and G. F. Schröder, Science, 2017, 358, 116–119 CrossRef PubMed.
- L. M. De Leon Rodriguez, Y. Hemar, J. Cornish and M. A. Brimble, Chem. Soc. Rev., 2016, 45, 4797–4824 RSC.
- B. Frohm, J. E. DeNizio, D. S. Lee, L. Gentile, U. Olsson, J. Malm, K. S. Akerfeldt and S. Linse, Soft Matter, 2015, 11, 414–421 RSC.
- G. M. Whitesides and M. Bocheva, Beyond molecules: self-assembly of mesoscopic and macroscopic components, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4769–4774 CrossRef PubMed.
- A. P. Schenning and E. W. Meijer, Chem. Commun., 2005, 3245–3258 RSC.
- J. Li, A. Corma and J. Yu, Chem. Soc. Rev., 2015, 44, 7112–7127 RSC.
- S. Garai, M. Rubčić, H. Bögge, E. T. Haupt, P. Gouzerh and A. Müller, Angew. Chem., Int. Ed., 2015, 54, 5879–5882 CrossRef PubMed.
- Y. Cui, Int. J. Pharm., 2007, 339, 3–18 CrossRef PubMed.
- J. C. Kendrew, G. Bodo, H. M. Dintzis, R. G. Parrish, H. Wyckoff and D. C. Phillips, Nature, 1958, 181, 662–666 CrossRef PubMed.
- B. V. Prasad and M. F. Schmid, Adv. Exp. Med. Biol., 2012, 726, 17–47 CrossRef PubMed.
- T. J. Mitchison, Philos. Trans. R. Soc., B, 1992, 336, 99–106 CrossRef PubMed.
- G. Sessa and G. Weissmann, J. Lipid Res., 1968, 9, 310–318 Search PubMed.
- G. Wei, Z. Su, N. P. Reynolds, P. Arosio, I. W. Hamley, E. Gazit and R. Mezzenga, Chem. Soc. Rev., 2017, 46, 4661–4708 RSC.
- T. P. Knowles and M. J. Buehler, Nat. Nanotechnol., 2011, 6, 469–479 CrossRef PubMed.
- T. P. Knowles and R. Mezzenga, Adv. Mater., 2016, 28, 6546–6561 CrossRef PubMed.
- T. P. Knowles, A. De Simone, A. W. Fitzpatrick, A. Baldwin, S. Meehan, L. Rajah, M. Vendruscolo, M. E. Welland, C. M. Dobson and E. M. Terentjev, Phys. Rev. Lett., 2012, 109, 158101 CrossRef PubMed.
- A. A. Chernov, J. Mater. Sci.: Mater. Electron., 2001, 12, 437–449 CrossRef.
- G. A. Garcia, S. I. A. Cohen, C. M. Dobson and T. P. J. Knowles, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2014, 89, 032712 CrossRef PubMed.
- B. Linse and S. Linse, Mol. BioSyst., 2011, 7, 2296–2303 RSC.
- N. S. Bieler, T. P. J. Knowles, D. Frenkel and R. Vácha, PLoS Comput. Biol., 2012, 8, e1002692 CrossRef PubMed.
- (a) A. Šarić, Y. C. Chebaro, T. P. J. Knowles and D. Frenkel, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17869–17874 CrossRef PubMed; (b) A. Šarić, T. C. T. Michaels, A. Zaccone, T. P. J. Knowles and D. Frenkel, J. Chem. Phys., 2016, 145, 211926 CrossRef PubMed.
- A. F. Wallace, L. O. Hedges, A. Fernandez-Martinez, P. Raiteri, J. D. Gale, G. A. Waychunas, S. Whitelam, J. F. Banfield and J. J. De Yoreo, Science, 2013, 341, 885–889 CrossRef PubMed.
- M. P. Hughes, M. R. Sawaya, D. R. Boyer, L. Goldschmidt, J. A. Rodriguez, D. Cascio, L. Chong, T. Gonen and D. S. Eisenberg, Science, 2018, 359, 698–701 CrossRef PubMed.
- (a) S. Ambadipudi, J. Biernat, D. Riedel, E. Madelkow and M. Zweckstetter, Nat. Commun., 2017, 17, 275 CrossRef PubMed; (b) S. Wegmann, B. Eftekharzadeh, K. Tepper, K. M. Zoltowska, R. E. Bennett, S. Dujardin, P. R. Laskowski, D. MacKenzie, T. Kamath, C. Commins, C. Vanderburg, A. D. Roe, Z. Fan, A. M. Molliex, A. Hernandez-Vega, D. Muller, A. A. Hyman, E. Mandelkow, J. P. Taylor and B. T. Hyman, EMBO J, 2018, 37, e98049 CrossRef PubMed.
- S. Qamar, G. Wang, S. J. Randle, F. S. Ruggeri, J. A. Varela, J. Q. Lin, E. C. PhillipsA. Miyashita, D. Williams, F. Ströhl, W. Meadows, R. Ferry, V. J. Dardov, G. G. Tartaglia, L. A. Farrer, G. S. Kaminski Schierle, C. F. Kaminski, C. E. Holt, P. E. Fraser, G. Schmitt-Ulms, D. Klenerman, T. Knowles, M. Vendruscolo and P. St George-Hyslop, Cell, 2018, 173, 720–734 CrossRef PubMed.
- H. A. Miers and P. Isaac, Chem. Soc. J., 1906, 89, 413–415 RSC.
- A. McPherson, Crystallization of Biological Macromolecules, CSHL Press, Cold Spring Harbor, 1999 Search PubMed.
- N. Asherie, Methods, 2004, 34, 266–272 CrossRef PubMed.
- L. Lindfors, S. Forssén, J. Westergren and U. Olsson, J. Colloid Interface Sci., 2008, 325, 404–413 CrossRef PubMed.
- M. J. Mullin, Crystallization, Butterworth-Heinemann, Oxford, 4th edn, 2001, ISBN: 9780750648332 Search PubMed.
- (a) S. Chia, P. Flagmeier, J. Habchi, V. Lattanzi, S. Linse, C. M. Dobson, T. P. J. Knowles and M. Vendruscolo, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 8005–8010 CrossRef PubMed; (b) C. Galvagnion, A. K. Buell, G. Meisl, T. C. Michaels, M. Vendruscolo, T. P. Knowles and C. M. Dobson, Nat. Chem. Biol., 2015, 11, 229–234 CrossRef PubMed.
- S. Campioni, G. Carret, S. Jordens, L. Nicoud, R. Mezzenga and R. Riek, J. Am. Chem. Soc., 2014, 136, 2866–2875 CrossRef PubMed.
- S. G. Agrawal and A. H. J. Paterson, Chem. Eng. Commun., 2015, 202, 98–706 CrossRef.
- G. D. Botsaris, Industrial Crystallization, Springer, 1976, pp. 3–22 Search PubMed.
- D. K. Kondepudi, R. J. Kaufman and N. Singh, Science, 1990, 250, 975–976 CrossRef PubMed.
- R. Y. Waghmare, J. N. Webb, T. W. Randolph, M. A. Larson and C. E. Glatz, J. Cryst. Growth, 2000, 208, 678–686 CrossRef.
- A. Srisa-nga, A. E. Flood and E. T. White, Cryst. Growth Des., 2006, 6, 795–801 CrossRef.
- S. Tait, E. T. White and J. D. Litster, Cryst. Growth Des., 2009, 9, 2198–2206 CrossRef.
- R. Chow, R. Blindt, R. Chivers and M. Povey, Ultrasonics, 2005, 43, 227–230 CrossRef PubMed.
- G. L. Gardner and G. K. Nancollas, J. Dent. Res., 1976, 55, 342–352 CrossRef PubMed.
- R. Zong, Y. Wang, S. Shi and Y. Zhu, Phys. Chem. Chem. Phys., 2014, 16, 4236–4241 RSC.
- P. Cubillas and M. W. Anderson, in Zeolites and Catalysis, Synthesis, Reactions and Applications, ed. A. Corma and S. Zones, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010, vol. 1, pp. 1–56 Search PubMed.
- C. S. Cundy and P. A. Cox, Microporous Mesoporous Mater., 2005, 82, 1–78 CrossRef.
- Z. L. Cheng, Z. Liu and S. Wang, J. Nanosci. Nanotechnol., 2016, 16, 1155–1159 CrossRef PubMed.
- J. Anwar, S. Khan and L. Lindfors, Angew. Chem., Int. Ed. Engl., 2015, 54, 14681–14684 CrossRef PubMed.
- J. Garside and R. J. Davey, Chem. Eng. Commun., 1980, 4, 393–424 CrossRef.
- E. Verdurand, C. Bebon, D. Colson, J.-P. Klein, A.-F. Blandin and J.-M. Bossoutrot, J. Cryst. Growth, 2005, 275, e1363–e1367 CrossRef.
- M. Bartkowiak, S. N. Fisher, A. M. Guénault, R. P. Haley, G. R. Pickett, G. N. Plenderleith and P. Skyba, Phys. Rev. Lett., 2000, 85, 4321–4324 CrossRef PubMed.
- F. A. Ferrone, J. Hofrichter, H. R. Sunshine and W. A. Eaton, Biophys. J., 1980, 32, 361–380 CrossRef PubMed.
- M. F. Bishop and F. A. Ferrone, Biophys. J., 1984, 46, 631–644 CrossRef PubMed.
- F. A. Ferrone, J. Hofrichter and W. A. Eaton, J. Mol. Biol., 1985, 183, 611–631 CrossRef PubMed.
- Y.-R. Qian and D. Botsaris, Chem. Eng. Sci., 1997, 52, 3429–3440 CrossRef.
- D. P. Kondepudi and K. Asakura, Acc. Chem. Res., 2001, 34, 946–954 CrossRef PubMed.
- M. O. Besenhard, P. Neugebauer, O. Scheibelhofer and J. G. Khinast, Cryst. Growth Des., 2017, 17, 6432–6444 CrossRef PubMed.
- Y.-R. Qian and D. Botsaris, Chem. Eng. Sci., 1998, 53, 1745–1756 CrossRef.
- E. G. Denk and G. D. Botsaris, J. Cryst. Growth, 1972, 13/14, 493–499 CrossRef.
- V. Foderà, F. Librizzi, M. Groenning, M. van de Weert and M. Leone, J. Phys. Chem. B, 2008, 112, 3853–3858 CrossRef PubMed.
- A. M. Ruschak and A. D. Miranker, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12341–12346 CrossRef PubMed.
- S. I. Cohen, S. Linse, L. M. Luheshi, E. Hellstrand, D. A. White, L. Rajah, D. E. Otzen, M. Vendruscolo, C. M. Dobson and T. P. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9758–9763 CrossRef PubMed.
- G. Meisl, X. Yang, E. Hellstrand, B. Frohm, J. B. Kirkegaard, S. I. Cohen, C. M. Dobson, S. Linse and T. P. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9384–9389 CrossRef PubMed.
- A. K. Buell, C. Galvagnion, R. Gaspar, E. Sparr, M. Vendruscolo, T. P. Knowles, S. Linse and C. M. Dobson, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7671–7676 CrossRef PubMed.
- R. Gaspar, G. Meisl., A. K. Buell, L. Young, C. F. Kaminski, T. P. J. Knowles, E. Sparr and S. Linse, Q. Rev. Biophys., 2017, 50, e6 CrossRef PubMed.
- D. K. Garg and B. Kundu, Biochim. Biophys. Acta, 2016, 1864, 794–804 CrossRef PubMed.
- T. P. Knowles, C. A. Waudby, G. L. Devlin, S. I. Cohen, A. Aguzzi, M. Vendruscolo, E. M. Terentjev, M. E. Welland and C. M. Dobson, Science, 2009, 326, 1533–1537 CrossRef PubMed.
- S. I. Cohen, M. Vendruscolo, M. E. Welland, C. M. Dobson, E. M. Terentjev and T. P. Knowles, J. Chem. Phys., 2011, 135, 065105 CrossRef PubMed.
- S. I. Cohen, M. Vendruscolo, C. M. Dobson and T. P. Knowles, J. Chem. Phys., 2011, 135, 065106 CrossRef PubMed.
- S. I. Cohen, M. Vendruscolo, C. M. Dobson and T. P. Knowles, J. Chem. Phys., 2011, 135, 065107 CrossRef PubMed.
- S. I. Cohen, M. Vendruscolo, C. M. Dobson and T. P. Knowles, J. Mol. Biol., 2012, 421, 160–171 CrossRef PubMed.
- P. Arosio, R. Cukalevski, B. Frohm, T. P. Knowles and S. Linse, J. Am. Chem. Soc., 2014, 136, 219–225 CrossRef PubMed.
- M. Grey, S. Linse, H. Nilsson, P. Brundin and E. Sparr, J. Parkinson's Dis., 2011, 1, 359–371 Search PubMed.
- R. Vácha, S. Linse and M. Lund, J. Am. Chem. Soc., 2014, 136, 11776–11782 CrossRef PubMed.
- S. I. A. Cohen, R. Cukalevski, T. C. T. Michaels, A. Saric, M. Törnquist, M. Vendruscolo, C. M. Dobson, A. K. Buell, T. P. J. Knowles and S. Linse, Nat. Chem., 2018, 10, 523–531 CrossRef PubMed.
- E. Roduner, Chem. Soc. Rev., 2014, 43, 8226–8239 RSC.
- G. Meisl, X. Yang, C. M. Dobson, S. Linse and T. P. J. Knowles, Chem. Sci., 2017, 8, 4352–4362 RSC.
- A. Abelein, J. Jarvet, A. Barth, A. Gräslund and J. Danielsson, J. Am. Chem. Soc., 2016, 138, 6893–6902 CrossRef PubMed.
- G. Meisl, X. Yang, B. Frohm, T. P. Knowles and S. Linse, Sci. Rep., 2016, 6, 18728 CrossRef PubMed.
- B. Bolognesi, S. I. Cohen, P. Aran Terol, E. K. Esbjörner, S. Giorgetti, M. F. Mossuto, A. Natalello, A. C. Brorsson, T. P. Knowles, C. M. Dobson and L. M. Luheshi, ACS Chem. Biol., 2014, 9, 378–382 CrossRef PubMed.
- X. Yang, G. Meisl, B. Frohm, E. Thulin, T. P. J. Knowles and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E5849–E5858 CrossRef PubMed.
- K. Eden, R. Morris, C. E. MacPhee and R. J. Allen, Biophys. J., 2015, 108, 632–643 CrossRef PubMed.
- A. Saric, A. Buell, G. Meisl, T. C. T. Michaels, C. Dobson, S. Linse, T. P. J. Knowles and D. Frenkel, Nat. Phys., 2016, 12, 874 Search PubMed.
- D. W. Li, S. A. Mohanty, A. Irbäck and S. Huo, PLoS Comput. Biol., 2008, 4, e1000238 CrossRef PubMed.
- F. Baftizadeh, F. Pietrucci, X. Biarnés and A. Laio, Phys. Rev. Lett., 2013, 110, 168103 CrossRef PubMed.
- J. Yang, A. J. Dear, T. C. T. Michaels, C. M. Dobson, T. P. J. Knowle, S. Wu and S. Perrett, J. Am. Chem. Soc., 2018, 140, 2493–2503 CrossRef PubMed.
- T. C. T. Michaels et al. , unpublished.
- D. M. Walsh, I. Klyubin, J. V. Fadeeva, W. K. Culle, R. Anwyl, M. S. Wolfe, M. J. Rowan and D. J. Selkoe, Nature, 2002, 416, 535–539 CrossRef PubMed.
- M. Bucciantini, E. Giannoni, F. Chiti, F. Baroni, L. Formigli, J. Zurdo, N. Taddei, G. Ramponi, C. M. Dobson and M. Stefani, Nature, 2002, 416, 507–511 CrossRef PubMed.
- C. Haass and D. J. Selkoe, Nat. Rev. Mol. Cell Biol., 2007, 8, 101–112 CrossRef PubMed.
- S. Baglioni, F. Casamenti, M. Bucciantini, L. M. Luheshi, N. Taddei, F. Chiti, C. M. Dobson and M. Stefani, J. Neurosci., 2006, 26, 8160–8167 CrossRef PubMed.
- A. Jan, O. Adolfsson, I. Allaman, A. L. Buccarello, P. J. Magistretti, A. Pfeifer, A. Muhs and H. A. Lashuel, J. Biol. Chem., 2011, 286, 8585–8596 CrossRef PubMed.
- S. I. Cohen, P. Arosio, J. Presto, F. R. Kurudenkandy, H. Biverstål, L. Dolfe, C. Dunning, X. Yang, B. Frohm, M. Vendruscolo, J. Johansson, C. M. Dobson, A. Fisahn, T. P. Knowles and S. Linse, Nat. Struct. Mol. Biol., 2015, 22, 207–213 CrossRef PubMed.
- K. W. Tipping, P. van Oosten-Hawle, E. W. Hewitt and S. E. Radford, Trends Biochem. Sci., 2015, 40, 719–727 CrossRef PubMed.
- P. Arosio, T. P. Knowles and S. Linse, Phys. Chem. Chem. Phys., 2015, 17, 7606–7618 RSC.
- P. Frawley, N. A. Mitchell, C. T. O’Ciardha and K. W. Hutton, Chem. Eng. Sci., 2012, 75, 183–197 CrossRef.
- C. J. Dunning, G. McGauran, K. Willén, G. K. Gouras, D. J. O'Connell and S. Linse, ACS Chem. Neurosci., 2016, 17, 161–170 CrossRef PubMed.
- J. Hardy and D. Allsop, Trends Pharmacol. Sci., 1991, 12, 383–388 CrossRef PubMed.
- J. A. Hardy and G. A. Higgins, Science, 1992, 256, 184–185 CrossRef PubMed.
- P. Arosio, T. C. Michaels, S. Linse, C. Månsson, C. Emanuelsson, J. Presto, J. Johansson, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Nat. Commun., 2016, 7, 10948 CrossRef PubMed.
- G. Meisl, J. B. Kirkegaard, P. Arosio, T. C. Michaels, M. Vendruscolo, C. M. Dobson, S. Linse and T. P. Knowles, Nat. Protoc., 2016, 11, 252–272 CrossRef PubMed.
- F. A. Aprile, P. Sormanni, M. Perni, P. Arosio, S. Linse, T. P. J. Knowles, C. M. Dobson and M. Vendruscolo, Sci. Adv., 2017, 3, e1700488 CrossRef PubMed.
- A. Munke, J. Persson, T. Weiffert, E. De Genst, G. Meisl, P. Arosio, A. Carnerup, C. M. Dobson, M. Vendruscolo, T. P. J. Knowles and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6444–6449 CrossRef PubMed.
- J. Habchi, S. Chia, R. Limbocker, B. Mannini, M. Ahn, M. Perni, O. Hansson, P. Arosio, J. R. Kumita, P. K. Challa, S. I. Cohen, S. Linse, C. M. Dobson, T. P. Knowles and M. Vendruscolo, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E200–E208 CrossRef PubMed.
- H. Liu, L. Yu, X. Dong and Y. SunD, J. Colloid Interface Sci., 2017, 491, 305 CrossRef PubMed.
- E. Bove-Fenderson, R. Urano, J. E. Straub and D. A. Harris, J. Biol. Chem., 2017, 292, 16858–16871 CrossRef PubMed.
- J. W. Brown, A. K. Buell, T. C. Michaels, G. Meisl, J. Carozza, P. Flagmeier, M. Vendruscolo, T. P. Knowles, C. M. Dobson and C. Galvagnion, Sci. Rep., 2016, 6, 36010 CrossRef PubMed.
- https://www.alzforum.org/mutations .
- E. Portelius, T. Lashley, A. Westerlund, R. Persson, N. C. Fox, K. Blennow, T. Revesz and H. Zetterberg, Neurodegener. Dis., 2015, 15, 50–57 CrossRef PubMed.
- A. T. Welzel, J. E. Maggio, G. M. Shankar, D. E. Walker, B. L. Ostaszewski, S. Li, I. Klyubin, M. J. Rowan, P. Seubert, D. M. Walsh and D. J. Selkoe, Biochemistry, 2014, 53, 3908–3921 CrossRef PubMed.
- O. Szczepankiewicz, B. Linse, G. Meisl, E. Thulin, B. Frohm, C. Sala Frigerio, M. T. Colvin, A. C. Jacavone, R. G. Griffin, T. P. Knowles, D. M. Walsh and S. Linse, J. Am. Chem. Soc., 2015, 137, 14673–14685 CrossRef PubMed.
- C. Dammers, M. Schwarten, A. K. Buell and D. Willbold, Chem. Sci., 2017, 8(7), 4996–5004 RSC.
- T. Weiffert et al. , unpublished.
- G. Di Fede, et al. , Science, 2009, 323, 1473–1477 CrossRef PubMed.
- K. Sanagavarapu et al. , unpublished.
- D. Thacker et al. , unpublished.
- A. Abelein, A. Gräslund and J. Danielsson, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5407–5412 CrossRef PubMed.
- R. Cukalevski, X. Yang, G. Meisl, U. Weininger, K. Bernfur, B. Frohm, T. P. J. Knowles and S. Linse, Chem. Sci., 2015, 6, 4215–4233 RSC.
- Y. Xiao, B. Ma, D. McElheny, S. Parthasarathy, F. Long, M. Hoshi, R. Nussinov and Y. Ishii, Nat. Struct. Mol. Biol., 2015, 22, 499–505 CrossRef PubMed.
- A. T. Petkova, et al., A structural model for Alzheimer's -amyloid fibrils based on experimental constraints from solid state NMR, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16742–16747 CrossRef PubMed.
- A. T. Petkova, W.-M. Yau and R. Tycko, Experimental Constraints on Quaternary Structure in Alzheimer's β-Amyloid Fibrils, Biochemistry, 2006, 45, 498–512 CrossRef PubMed.
- A. K. Paravastu, R. D. Leapman, W.-M. Yau and R. Tycko, Molecular structural basis for polymorphism in Alzheimer's-amyloid fibrils, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18349–18354 CrossRef PubMed.
- I. Bertini, L. Gonnelli, C. Luchinat, J. Mao and A. Nesi, A New Structural Model of Aβ40 Fibrils, J. Am. Chem. Soc., 2011, 133, 16013–16022 CrossRef PubMed.
- B. K. Yoo, Y. Xiao, D. McElheny and Y. Ishii, J. Am. Chem. Soc., 2018, 140, 2781–2784 CrossRef PubMed.
- C. Condello, T. Lemmin, J. Stöhr, M. Nick, Y. Wu, A. M. Maxwell, J. C. Watts, C. D. Caro, A. Oehler, C. D. Keene, T. D. Bird, S. G. van Duinen, L. Lannfelt, M. Ingelsson, C. Graff, K. Giles, W. F. DeGrado and S. B. Prusiner, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E782–E791 CrossRef PubMed.
- R. Frankel et al. , unpublished.
- D. J. Lindberg, E. Wesén, J. Björkeroth, S. Rocha and E. K. Esbjörner, Biochim. Biophys. Acta, 2017, 1859, 1921–1929 CrossRef PubMed.
- J. Habchi, S. Chia, C. Galvagnion, T. C. T. Michales, M. M. J. Bellaiche, F. S. Ruggeri, M. Sanguanini, I. Idini, J. R. Kumita, E. Sparr, S. Linse, C. M. Dobson, T. P. J. Knowles and M. Vendruscolo, Nat. Chem., 2018, 10, 673–683 CrossRef PubMed.
- C. Galvagnion, J. Parkinson's Dis., 2017, 7, 433–450 Search PubMed.
- E. Hellstrand, E. Sparr and S. Linse, Biophys. J., 2010, 98, 2206–2214 CrossRef PubMed.
- (a) M. D. Kane, W. J. Lipinski, M. J. Callahan, F. Bian, R. A. Durham, R. D. Schwarz, A. E. Roher and L. C. Walker, J. Neurosci., 2000, 20, 3606–3611 CrossRef PubMed; (b) M. Meyer-Luehmann, J. Coomaraswamy, T. Bolmont, S. Kaeser, C. Schaefer, E. Kilger, A. Neuenschwander, D. Abramowski, P. Frey, A. L. Jaton, J. M. Vigouret, P. Paganetti, D. M. Walsh, P. M. Mathews, J. Ghiso, M. Staufenbiel, L. C. Walker and M. Jucker, Science, 2006, 313, 1781–1784 CrossRef PubMed.
- Y. S. Eisele, T. Bolmont, M. Heikenwalder, F. Langer, L. H. Jacobson, Z. X. Yan, K. Roth, A. Aguzz, M. Staufenbiel, L. C. Walker and M. Jucker, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 12926–12931 CrossRef PubMed.
- R. F. Rosen, J. J. Fritz, J. Dooyema, A. F. Cintron, T. Hamaguchi, J. J. Lah, H. LeVine 3rd, M. Jucker and L. C. Walker, J. Neurochem., 2012, 120, 660–666 CrossRef PubMed.
- R. Morales, J. Bravo-Alegria, C. Duran-Aniotz and C. Soto, Sci. Rep., 2015, 5, 9349 CrossRef PubMed.
- R. M. Ridley, Very long term studies of the seeding of β-amyloidosis in primates, J. Neural Transm., 2006, 113, 1243–1251 CrossRef PubMed.
- Z. Jaunmuktane, S. Mead, M. Ellis, J. D. F. Wadsworth, A. J. Nicoll, J. Kenny, F. Launchbury, J. Linehan, A. Richard-Loendt, A. S. Walker, P. Rudge, J. Collinge and S. Brandner, Nature, 2015, 525, 247–250 CrossRef PubMed.
- D. L. Ritchie, P. Adlard, A. H. Peden, S. Lowrie, M. Le Grice, K. Burns, R. J. Jackson, H. Yull, M. J. Keogh, W. Wei, P. F. Chinnery, M. W. Head and J. W. Ironside, Acta Neuropathol., 2017, 134, 221–240 CrossRef PubMed.
- A. Irbäck, S. Æ. Jónsson, N. Linnemann, B. Linse and S. Wallin, Phys. Rev. Lett., 2013, 110, 058101 CrossRef PubMed.
- T. T. Tran, P. H. Nguyen and P. Derreumaux, J. Chem. Phys., 2016, 144, 205103 CrossRef PubMed.
- F. Sterpone, S. Doutreligne, T. T. Tran, S. Melchionna, M. Baaden, P. H. Nguyen and P. Derreumaux, Biochem. Biophys. Res. Commun., 2018, 498, 296–304 CrossRef PubMed.
- M. Chiricotto, F. Sterpone, P. Derreumaux and S. Melchionna, Philos. Trans. R. Soc., A, 2016, 374, 20160225 CrossRef PubMed.
- S. Sasmal, N. Schwierz and T. Head-Gordon, J. Phys. Chem. B, 2016, 120, 12088–12097 CrossRef PubMed.
- N. Schwierz, C. V. Frost, P. L. Geissler and M. Zacharias, J. Phys. Chem. B, 2017, 121, 671–682 CrossRef PubMed.
- J. Nasica-Labouze, P. H. Nguyen, F. Sterpone, O. Berthoumieu, N. V. Buchete, S. Coté, A. De Simone, A. J. Doig, P. Faller, A. Garcia, A. Laio, M. S. Li, S. Melchionna, N. Mousseau, Y. Mu, A. Paravastu, S. Pasquali, D. J. Rosenman, B. Strodel, B. Tarus, J. H. Viles, T. Zhang, C. Wang and P. Derreumaux, Chem. Rev., 2015, 115, 3518–3563 CrossRef PubMed.
- B. Barz and B. Strodel, Chemistry, 2016, 22, 8768–8772 CrossRef PubMed.
- M. M. J. Bellaiche et al. , unpublished.
- E. Hellstrand, R. Boland, D. M. Walsh and S. Linse, ACS Chem. Neurosci., 2010, 1, 13–18 CrossRef PubMed.
- D. Walsh, E. Thulin, A. M. Minogue, N. Gustavsson, E. Pang, D. B. Teplow and S. Linse, FEBS J., 2009, 276, 1266–1281 CrossRef PubMed.
- The IUPAC Gold book, http://goldbook.iupac.org/html/M/M03804.html.
- J. S. Jeong, A. Ansaloni, R. Mezzenga, H. A. Lashuel and G. Dietler, J. Mol. Biol., 2013, 425, 1765–1781 CrossRef PubMed.
- M. Törnquist et al. , unpublished.
- A. W. Fitzpatrick, G. T. Debelouchina, M. J. Bayro, D. K. Clare, M. A. Caporini, V. S. Bajaj, C. P. Jaroniec, L. Wang, V. Ladizhansky, S. A. Müller, C. E. MacPhee, C. A. Waudby, H. R. Mott, A. De Simone, T. P. Knowles, H. R. Saibil, M. Vendruscolo, E. V. Orlova, R. G. Griffin and C. M. Dobson, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 5468–5473 CrossRef PubMed.
- L. E. Orgel, Chem. Biol., 1996, 3, 413–414 CrossRef PubMed.
- W. P. Esler, E. R. Stimson, J. B. Fishman, J. R. Ghilardi, H. V. Vinters, P. W. Mantyh and J. E. Maggio, Biopolymers, 1999, 49, 505–514 CrossRef PubMed.
- V. Torbeev, M. Grogg, J. Ruiz, R. Boehringer, A. Schirer, P. Hellwig, G. Jeschk and D. Hilvert, J. Pept. Sci., 2016, 22, 290–304 CrossRef PubMed.
- T. Ban, M. Hoshino, S. Takahashi, D. Hamada, K. Hasegawa, H. Naiki and Y. Goto, Direct Observation of Aβ Amyloid Fibril Growth and Inhibition, J. Mol. Biol., 2004, 344, 757–767 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |