
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organic photocatalysis for the radical couplings of boronic acid derivatives in batch and flow†
Fabio
Lima
 a,
Lars
Grunenberg‡
a,
Husaini B. A.
Rahman‡
a,
Ricardo
Labes
a,
Joerg
Sedelmeier
b and
Steven V.
Ley
*a
a,
Lars
Grunenberg‡
a,
Husaini B. A.
Rahman‡
a,
Ricardo
Labes
a,
Joerg
Sedelmeier
b and
Steven V.
Ley
*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: svl1000@cam.ac.uk; Web: http://www.leygroup.ch.cam.ac.uk
bNovartis Pharma AG, Novartis Campus, 4002 Basel, Switzerland
First published on 16th May 2018
Abstract
We report an acridium-based organic photocatalyst as an efficient replacement for iridium-based photocatalysts to oxidise boronic acid derivatives by a single electron process. Furthermore, we applied the developed catalytic system to the synthesis of four active pharmaceutical ingredients (APIs). A straightforward scale up approach using continuous flow photoreactors is also reported affording gram an hour throughput.
Visible-light photoredox catalysis has become an important tool for radical species generation without the need for stoichiometric reagents.1 Using these methods, a large number of applications have emerged for the catalytic generation of carbon radical species via single electron oxidation of charged anionic species, such as carboxylates, silicates, and trifluoroborate salts.2 We recently developed a photoredox method to generate radicals from neutral boronic acids or esters, making use of an additional Lewis base catalyst (LB) to activate the trivalent boronic species towards single-electron oxidation.3 The substantial number of compatible substrates, combined with their commercial availability makes it a method of choice to generate carbon radicals catalytically and engage them in versatile radical-based coupling reactions. However, effective oxidation of these redox-active complexes has until now only been realised using costly iridium based photoredox catalysts (Ir-1 or Ir-2). Despite the promises offered by the photoredox disconnections for pharmaceutical products synthesis,4 industry has to comply with residual transition metal limits in final products (oral permitted daily exposure for iridium ≤100 μg per day).5 To comply with these low residual metal levels, costly purification protocols have to be carried out after transformations using transition metal catalysts. With the recent development of efficient new generation organic photocatalysts,5a,6 we sought to identify a suitable organic replacement to the previously used iridium photosensitisers.7 Ideally this protocol could be scaled up using a continuous flow reactor and its utility demonstrated by synthesising pharmaceutically relevant compounds in an efficient manner.
To start this investigation, we chose a model radical conjugate addition reaction of boronic ester 1 or boronic acid 2 with methyl vinyl ketone (MVK, 3) as radical acceptor.3b We subjected these species to a series of photoredox catalysts in the presence of catalytic amount of Lewis base (LB = quinuclidin-3-ol or DMAP) and under blue LEDs (14 W at λ = 450 nm) irradiation to produce the coupled products 4 or 5 (Scheme 1). A wide range of visible-light absorbing organic photocatalysts were screened using these two model reactions (full table in ESI†).
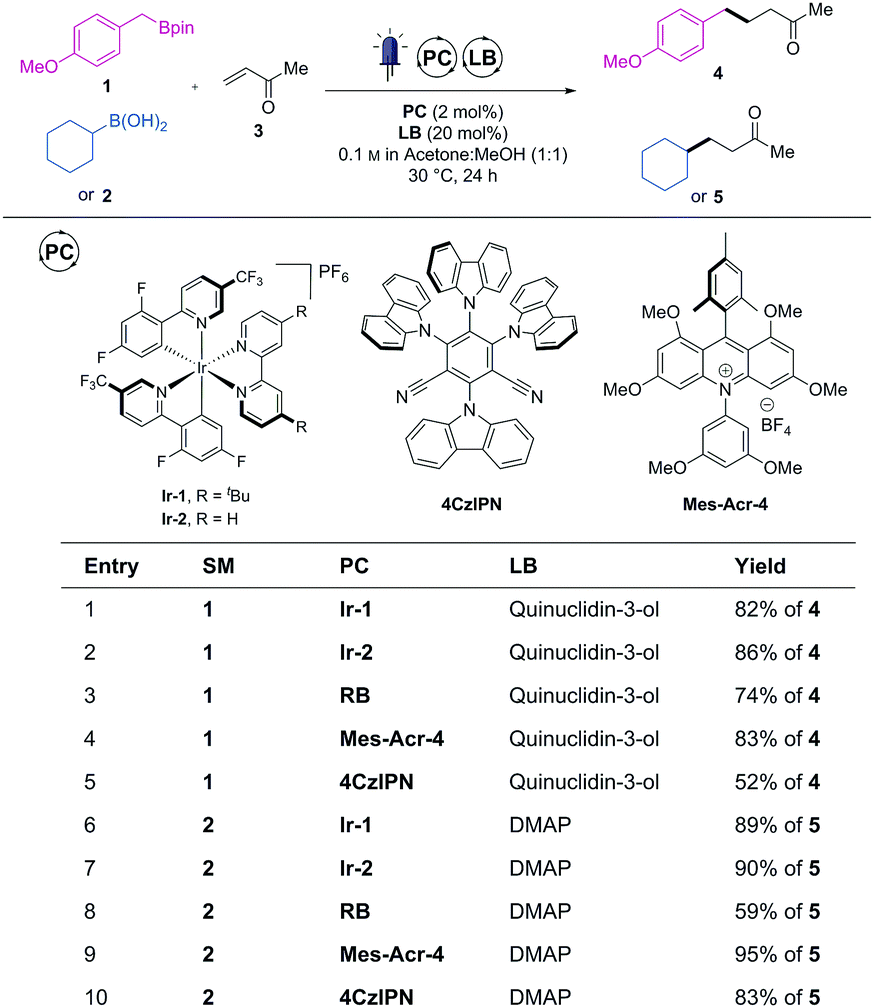 | ||
| Scheme 1 Selected examples of catalyst performance. See ESI† for full optimisation. | ||
Along with rose bengal (RB), two recently developed organic dyes (4CzlPN8 and Mes-Acr-46) emerged from this screening as potential organic replacement to Ir-1 and Ir-2. While organic dyes degradation upon prolonged irradiation can be a potential pitfall, no discolouration (or photobleaching) occurred during the course of the irradiation with these three photocatalysts. It was therefore postulated that these dyes were stable enough to be used without decline of reactivity. Finally, as Mes-Acr-4 consistently led to the highest yields (similar to Ir-1 and Ir-2) with both the boronic ester (1, entry 4) and acid (2, entry 9), it was chosen as a promising replacement to the iridium-based photocatalysts.
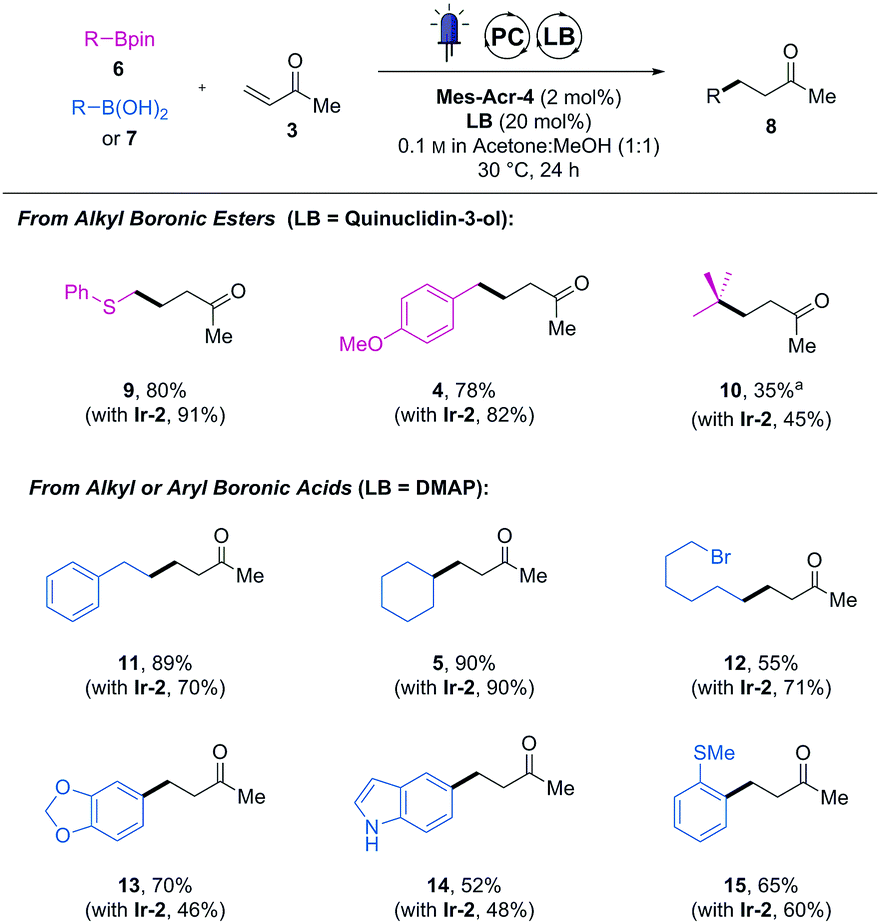
We then further compared the generality of Mes-Acr-4 against Ir-2 with a wider range boronic acid derivatives (Table 1). As far as the cross-coupled product yields are concerned, the performance of the Mes-Acr-4 dye was often equivalent to the Ir-2 photocatalyst.
| Isolated yields. Reactions carried out with 0.2 mmol of 6 or 7, 0.8 mmol of 3, 2 mol% of Mes-Acr-4 and 20 mol% of the indicated Lewis base (LB).a DMAP was used as a Lewis base. Isolated yield obtained with Ir-2 for the same product in parentheses. |
|---|
|
While products 4, 5 and 14 were obtained in the same isolated yields with both photocatalysts, the organic dye was superior to the iridium one for reactions leading to 11, 13 and 15. However, in some cases, inferior yields were observed with the organic dye (see compounds 9, 10 and 12). Although no apparent correlation between the structure of the boronic acid derivative and their reactivity could be drawn, no reactivity shutdown was observed by replacing the iridium-photocatalyst by Mes-Acr-4.
To more finely compare the two catalysts, a reaction profile of the conjugate addition of the cyclohexyl boronic acid (2) was recorded by following the reaction using NMR spectroscopy (Fig. 1). To do this, reagents were dissolved in a deuterated solvent mixture and irradiated inside a borosilicate NMR tube. At different time points, the light was turned off, thereby putting the reaction on hold to conduct a 1H-NMR measurement. Using 1,3,5-trimethoxy benzene as an internal standard, it was possible to determine the yield in the deuterated product (5d) over time. Using this method, we were able to conveniently monitor the reaction progress with minimal material usage (0.05 mmol of 2).
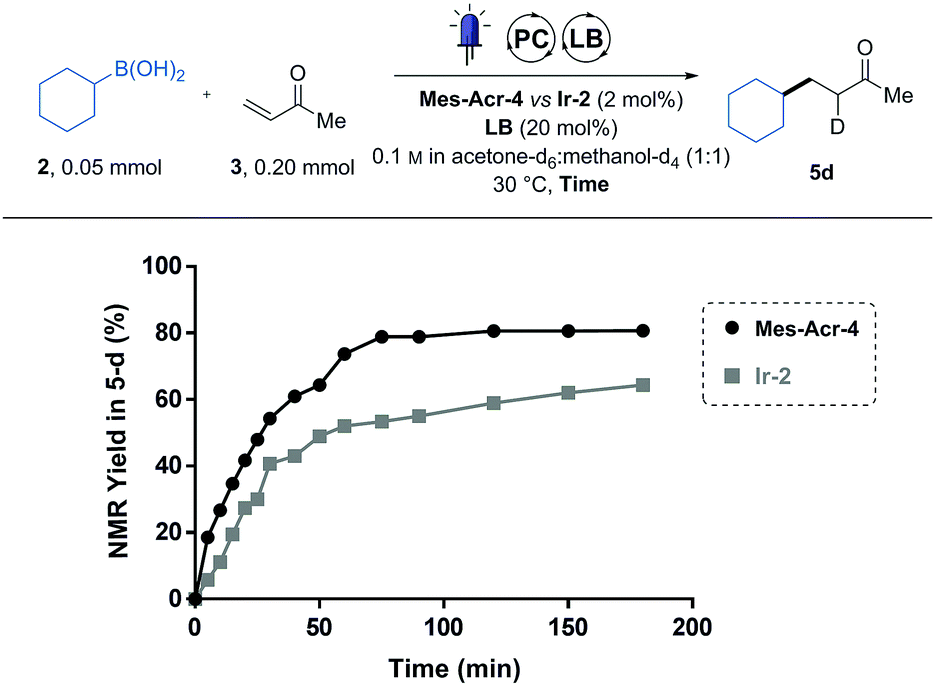 | ||
| Fig. 1 Reaction following of Mes-Acr-4 against Ir-2 in NMR tube. Reaction monitoring using 1H-NMR, comparison between Mes-Ar-4 (dots) and Ir-2 (squares). Reactions carried out at 0.05 mmol of 2, 0.2 mmol of 3, 2 mol% of the indicated PC and 20 mol% DMAP in deuterated solvents. | ||
While the two catalysts performed equally after the 24 h of irradiation (5 in Table 1), we could observe that they do not share the same reaction profile (Fig. 1). Although the two curves seem to follow similar initial kinetics, the slope of the Ir-2 curve suddenly drops after 30 min while the Mes-Acr-4 one continues to follow a more regular path. We postulated that this behaviour is a sign of a catalyst deactivation pathway.9 Indeed, radical alkylation of the pyridyl ligands has already been observed on similar iridium-based photoredox catalysts.9 Also, 2,2′-bipyridyl (bpy) ligand substitution (of Ir-2) in acetonitrile solvent was identified as another deactivation pathway for these photocatalysts.10 We therefore postulated that the strongly nucleophilic DMAP catalyst can result in bpy displacement on the iridium centre of Ir-2 and lead to catalyst deactivation. DiRocco demonstrated that the Mes-Acr-4 catalyst was reasonably stable under similar conditions.6 From this analysis we discovered that the Mes-Acr-4 catalyst brings faster kinetics than Ir-2 at the same catalyst loading and is therefore more effective for this reaction.
Pleasingly, this reaction monitoring also informed us that the reaction can be completed in 70 min, with this new catalyst. This relatively short reaction time, combined with the benefits of microscale on photoreactions,11 motivated us to study the transferability of this process in a continuous flow reactor. To facilitate this transfer, we initially compared the reaction profile observed in the NMR tube (14 W at λ = 450 nm) to the yields obtained in a Vapourtec UV-150 reactor equipped with blue LEDs (17 W at λ = 420 nm). These reactions were performed individually with different residence times, and the kinetic profile proved to be similar (Fig. 2). While observed kinetics for photoreactions limited by photon-flux are often significantly different in a large batch vessel than in the small channels of a photo flow reactor,11a we observed similar yields after the same time of irradiation between the two systems.
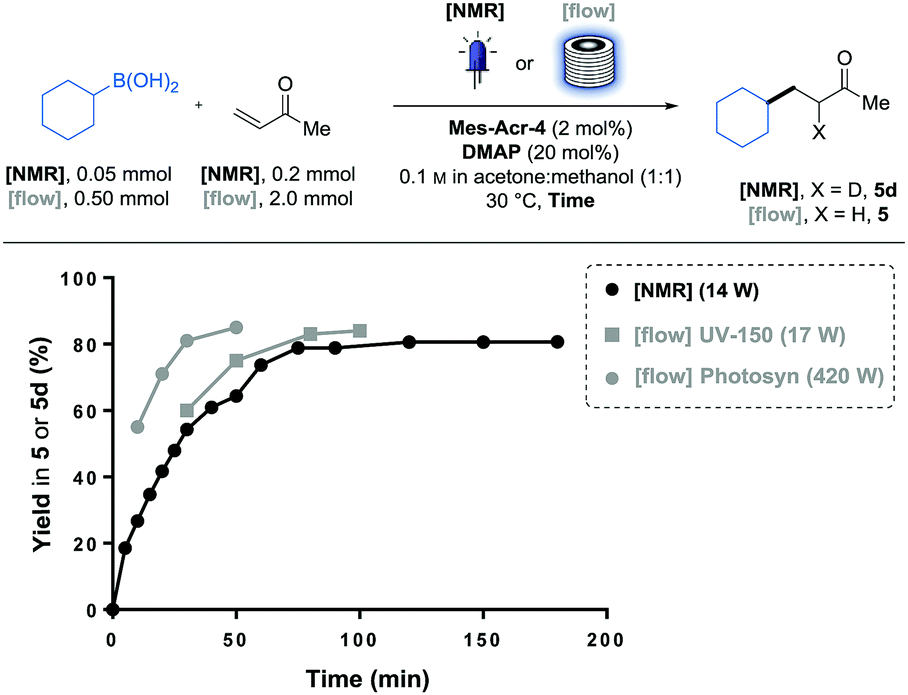 | ||
| Fig. 2 Yield comparison between NMR tube and flow irradiation. Comparison between batch (black) and flow (grey) modes. UV-150 (10 mL FEP coil) and Photosyn (5 mL PFA coil). | ||
We hypothesised that the inner diameter (i.d.) of the NMR tube (i.d. = 5.0 mm) was not large enough compared to the FEP tubing of the UV-150 coil (i.d. = 1.3 mm) to observe a significant difference in terms of light absorption efficiency. With these reaction conditions and using reported data on similar photocatalysts,11c approximately 90% of the light is absorbed within the first 0.3 mm of the solution. Reaction rate could also be dependent on the mass transfer.11b Nevertheless, as the reaction following curve obtained via NMR matched with the kinetics observed in the UV-150 photoreactor (10 mL), we decided to use the NMR reaction monitoring to predict the residence time required to reach the full conversion in the flow photoreactor. As every substrate will require a different irradiation time, we will be able to circumvent the material- and time-consuming residence time optimisation on the flow equipment by simulating the required irradiation time with a single batch experiment.12
To complete this flow photoreactor investigation, we examined this model reaction in a photoreactor (Photosyn) recently developed by Uniqsis Ltd and that was made available in our lab for a brief testing period. The powerful LEDs (420 W at λ = 450 nm) equipped on this reactor allowed faster rates to be observed in a 5 mL investigation coil (PFA, i.d. = 1.0 mm). This massive power increase brought the irradiation time necessary to reach 80% yield of 5 from 70 min in the UV-150 reactor to 30 min in the Photosyn. This significant rate enhancement encouraged us to run a larger scale experiment on a 50 mL coil (PFA, i.d. = 2.4 mm) using the same irradiation system (Scheme 2). Pleasingly the results obtained in the 5 mL coil translated well in the 50 mL coil in which we could obtain 3.7 g (24.1 mmol, 81% yield) of 5 in 3 h, corresponding to a throughput of 8.0 mmol h−1.
 | ||
Scheme 2 Larger scale reaction in photosyn (420 W at λ = 450 nm). Reaction carried out at 0.1 M concentration of 2 in acetone![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol (1:1) using 2 mol% of Mes-Acr-4 and 20 mol% of DMAP. :methanol (1:1) using 2 mol% of Mes-Acr-4 and 20 mol% of DMAP. | ||
With a robust organocatalytic system in hand to activate boronic acid derivatives and transfer the chemistry in a flow photoreactor, we decided to explore its application for effective active pharmaceutical ingredients synthesis.4a As powerful approaches using photogenerated α-amino radicals were reported for the concise synthesis of baclofen13 and pregabalin,14 we were therefore naturally inclined to test the applicability of the recently developed methodology for the synthesis of active pharmaceutical ingredients (API) from the γ-amino butyric acid (GABA) family.
This approach uses the Boc-protected amino boronic ester (16) as the α-amino radical source. This substrate would undergo our recently developed Lewis base and photoredox dual catalysed addition to diethyl malonate-derived olefins (17).3b The resulting coupled product (18) could then be deprotected, hydrolysed and decarboxylated using aqueous HCl (6 M) and heating to reveal the amino acid hydrochlorides (19).13 We initially studied the feasibility of this transformation in batch (Table 2). After a brief optimisation of the reaction conditions, we could couple the methyl-amino building block (16) to malonate derived trisubstituted olefins in high yields (20 to 22). A further excess of 16 and longer irradiation time (48 h instead of 18 h) was required to couple the tetra-substituted olefin in acceptable yield (23).
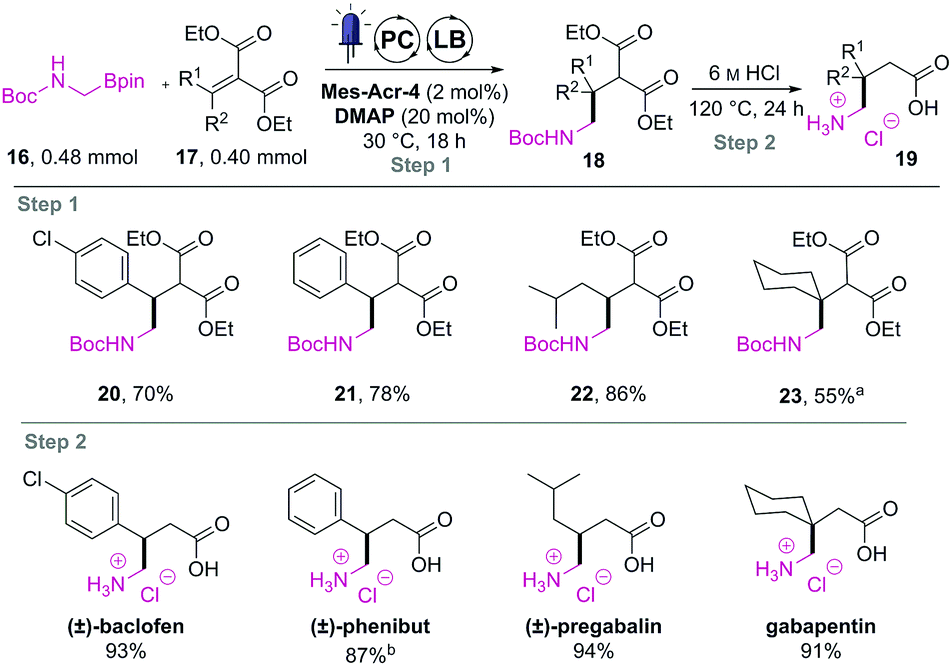
The isolated drug precursors were then subjected to one pot deprotection, hydrolysis, and decarboxylation using aqueous HCl. While phenibut was obtained without residual lactam formation after only 1 h of reflux, the other precursors required prolonged heating (24 h) to fully hydrolyse their corresponding γ-lactams to the final crystalline APIs as hydrochloride salts in high yields. With these encouraging results, we decided to apply this method in the flow synthesis of these active pharmaceutical ingredients. Tests on telescoping the hydrolysis of 18 revealed that the impurity profile carried out from step 1 would make the purity of the final APIs insufficient to crystallise.15 Therefore, only the photoredox coupling step was investigated in a flow reactor.16 As observed during the optimisation study, the long irradiation time required for the synthesis of the gabapentin precursor (23) prevented us from transferring this reaction to flow (τ = 100 min limit).
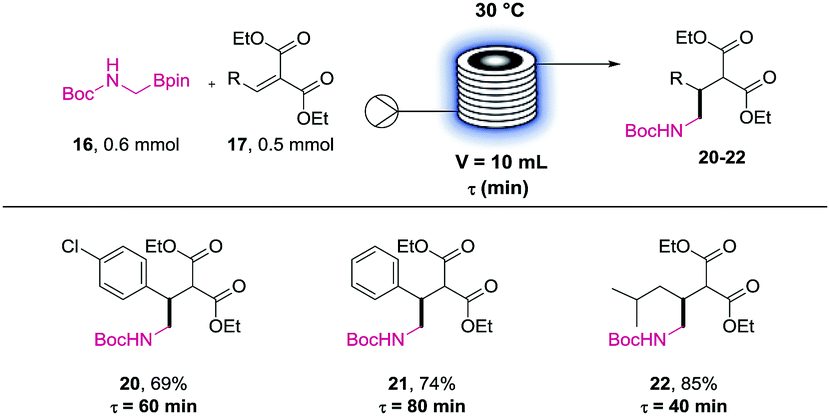
Therefore, we investigated only the reaction profile of the three other precursors (20 to 22) using the NMR method previously described (see ESI†). These profiles allowed us to identify the residence time needed for each partner. While the deuterated phenibut precursor required 80 min of irradiation, the baclofen and pregabalin ones could be prepared efficiently in 60 min and 40 min respectively.
Using these “predicted” residence times in the UV-150 setup resulted in finely tuned conditions for these APIs precursors coupling reactions (Table 3). Pleasingly, all the targeted compounds were successfully obtained in high yields after purification (20 to 22). These experiments confirm the feasibility of the photoredox coupling step in a flow reactor for potential scale up in a larger photoreactor.
| Isolated yields. 0.5 mmol scale reaction in UV-150 (17 W at λ = 420 nm). Reaction carried out at 0.1 M concentration of 17 in acetone:methanol (1:1) using 2 mol% of Mes-Acr-4 and 20 mol% of DMAP. |
|---|
|
As a summary, we identified the organic dye Mes-Acr-4 as an alternative to iridium photocatalysts for the photoredox activation of boronic acid derivatives. This organic photocatalyst have shown similar to better activity than Ir-2 in photoredox couplings with methyl vinyl ketone as well as enhanced stability under the reaction conditions. NMR reaction monitoring allowed us to rapidly predict the necessary residence time to transfer these reactions in flow. Gram per hour throughput could be obtained using a recently developed larger (50 mL) visible light photoreactor. The synthetic utility of this methodology could be further demonstrated by the two-step syntheses of four approved central nervous system drugs from the GABA family in batch and flow modes.
We are grateful to Novartis Pharma AG (F. L.), Erasmus Scholarship Scheme (L. G.) and the EPSRC (S. V. L., Grant No. EP/K009494/1, EP/K039520/1 and EP/M004120/1) for financial support. We thank Dr Berthold Schenkel and Dr Gottfried Sedelmeier form Novartis for insightful discussions. We thank Dr Mark Ladlow (UNIQSIS Ltd, UK) for the loan of the large scale Photosyn reactor.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J.-P. Goddard, C. Ollivier and L. Fensterbank, Acc. Chem. Res., 2016, 49, 1924–1936 CrossRef PubMed.
- (a) J. Xuan, Z. G. Zhang and W. J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632–15641 CrossRef PubMed; (b) V. Corcé, L. Chamoreau, E. Derat, J. Goddard, C. Ollivier and L. Fensterbank, Angew. Chem., Int. Ed., 2015, 54, 11414–11418 CrossRef PubMed; (c) Y. Yasu, T. Koike and M. Akita, Adv. Synth. Catal., 2012, 354, 3414–3420 CrossRef.
- (a) F. Lima, M. A. Kabeshov, D. N. Tran, C. Battilocchio, J. Sedelmeier, G. Sedelmeier, B. Schenkel and S. V. Ley, Angew. Chem., Int. Ed., 2016, 55, 14085–14089 CrossRef PubMed; (b) F. Lima, U. K. Sharma, L. Grunenberg, D. Saha, S. Johannsen, J. Sedelmeier, E. V. Van der Eycken and S. V. Ley, Angew. Chem., Int. Ed., 2017, 56, 15136–15140 CrossRef PubMed.
- (a) J. J. Douglas, M. J. Sevrin and C. R. J. Stephenson, Org. Process Res. Dev., 2016, 20, 1134–1147 CrossRef; (b) J. J. Douglas, K. P. Cole and C. R. J. Stephenson, J. Org. Chem., 2014, 79, 11631–11643 CrossRef PubMed.
- (a) M. Majek and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2015, 54, 2270–2274 CrossRef PubMed; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef PubMed; (c) N. G. Anderson, Practical Process Research and Development: A Guide for Organic Chemists, Academic Press, Oxford, 2012 Search PubMed.
- A. Joshi-Pangu, F. Lévesque, H. G. Roth, S. F. Oliver, L. C. Campeau, D. Nicewicz and D. A. DiRocco, J. Org. Chem., 2016, 81, 7244–7249 CrossRef PubMed.
- K. Teegardin, J. I. Day, J. Chan and J. Weaver, Org. Process Res. Dev., 2016, 20, 1156–1163 CrossRef PubMed.
- (a) C. Lévêque, L. Chenneberg, V. Corcé, C. Ollivier and L. Fensterbank, Chem. Commun., 2016, 52, 9877–9880 RSC; (b) J. Luo and J. Zhang, ACS Catal., 2016, 6, 873–877 CrossRef.
- J. J. Devery III, J. J. Douglas, J. D. Nguyen, K. P. Cole, R. A. Flowers II and C. R. J. Stephenson, Chem. Sci., 2015, 6, 537–541 RSC.
- I. N. Mills, J. A. Porras and S. Bernhard, Acc. Chem. Res., 2018, 51, 352–364 CrossRef PubMed.
- (a) D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed; (b) K. Loubière, M. Oelgemöller, T. Aillet, O. Dechy-Cabaret and L. Prat, Chem. Eng. Process., 2016, 104, 120–132 CrossRef; (c) Y. Su, N. J. W. Straathof, V. Hessel and T. Noël, Chem. – Eur. J., 2014, 20, 10562–10589 CrossRef PubMed.
- L. D. Elliott, J. P. Knowles, C. S. Stacey, D. J. Klauber and K. I. Booker-Milburn, React. Chem. Eng., 2018, 3, 86–93 Search PubMed.
- K. Miyazawa, T. Koike and M. Akita, Adv. Synth. Catal., 2014, 356, 2749–2755 CrossRef.
- L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886–10889 CrossRef PubMed.
- H. Ishitani, K. Kanai, Y. Saito, T. Tsubogo and S. Kobayashi, Eur. J. Org. Chem., 2017, 6491–6494 CrossRef.
- D. Ghislieri, K. Gilmore and P. H. Seeberger, Angew. Chem., Int. Ed., 2015, 54, 678–682 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc02169d |
| ‡ These authors contributed equally to the project. |
| This journal is © The Royal Society of Chemistry 2018 |