
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Small bite-angle 2-phosphinophosphinine ligands enable rhodium-catalysed hydroboration of carbonyls†
Robert J.
Newland
 a,
Jason M.
Lynam
b and
Stephen M.
Mansell
*a
a,
Jason M.
Lynam
b and
Stephen M.
Mansell
*a
aInstitute of Chemical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS, UK. E-mail: s.mansell@hw.ac.uk; Web: http://www.mansellresearch.org.uk
bDepartment of Chemistry, University of York, Heslington, York, YO10 5DD, UK
First published on 14th May 2018
Abstract
Two Rh complexes of the 2-phosphinophosphinine ligand 2-PPh2-3-Me-6-SiMe3-C5H2P (1) were prepared: dinuclear trans-[{Rh(CO)(Cl)(μ-1)}2] (2) and chelating [Rh(1)(COD)][B(ArF)4] (3). Despite the widespread use of Rh catalysts for the hydroboration of alkenes, 3 is reported to be the first Rh catalyst for ketone and ketimine hydroboration, with high activity observed at 0.1 mol% loading.
The catalytic hydroboration of carbonyl substrates1 is of current interest due to the importance of the controlled reduction of carbonyls to alcohols under mild conditions and the considerable safety advantages over the use of stoichiometric metal hydrides and catalytic hydrogenation.2 Transfer hydrogenation is an alternative reaction for the reduction of ketones, however, many catalysts require forcing conditions to achieve acceptable conversion.3 There has been a great deal of interest in developing catalysts for the hydroboration of carbonyl compounds, with catalysts based on s-block (e.g. Li,4 Na,5 Mg6) and p-block (e.g. B,7 Al,8 Ge, Sn9) elements, as well as first-row (e.g. Ti,10 Mn,11 Fe,12 Ni13), group 6 (Mo14) and late (Re,15 Ru16) transition metals recently reported. Despite the prevalence of Rh catalysts for the hydroboration of alkenes,17 to the best of our knowledge, there has been only one stoichiometric example of the use of a Rh complex in the hydroboration of a carbonyl compound (benzaldehyde) reported in the literature to date.18
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ‡ Based on the wide variety of hydroboration catalysts, and the wide-spread use of Rh catalysts in alkene hydroboration, its absence in carbonyl hydroboration was unexpected.
‡ Based on the wide variety of hydroboration catalysts, and the wide-spread use of Rh catalysts in alkene hydroboration, its absence in carbonyl hydroboration was unexpected.
The use of phosphinine (the P-analogue of pyridine) ligands in catalysis is a growing field19 with many recent contributions by Müller and co-workers in particular.20 With regards to Rh catalysis, Breit developed the use of mono- and multidentate phosphinine ligands for hydroformylation catalysis that highlighted several advantages of these unusual ligand systems in an industrially valuable reaction over classical ligands such as PPh3.21 Small bite-angle ligands have become an increasingly popular choice in catalysis,22 and our interests lie in the development of catalysts using small bite-angle 2-phosphinophosphinine ligands (Scheme 1, 1) due to their unique properties.19 In particular, the increased s-character (ca. 61%)23 of the formally sp2 phosphorus atom can lead to less-strained four-membered chelates. Evidence for this was observed in the chelating Ru complex cis-[Ru(1)2(Cl)2] (4),24 as well as in a series of κ2 group 6 tetracarbonyl complexes.25 With the increasing popularity of small bite-angle ligands in homogeneous catalysis,22 developing ligands that are less likely to form bridging architectures is highly desirable.
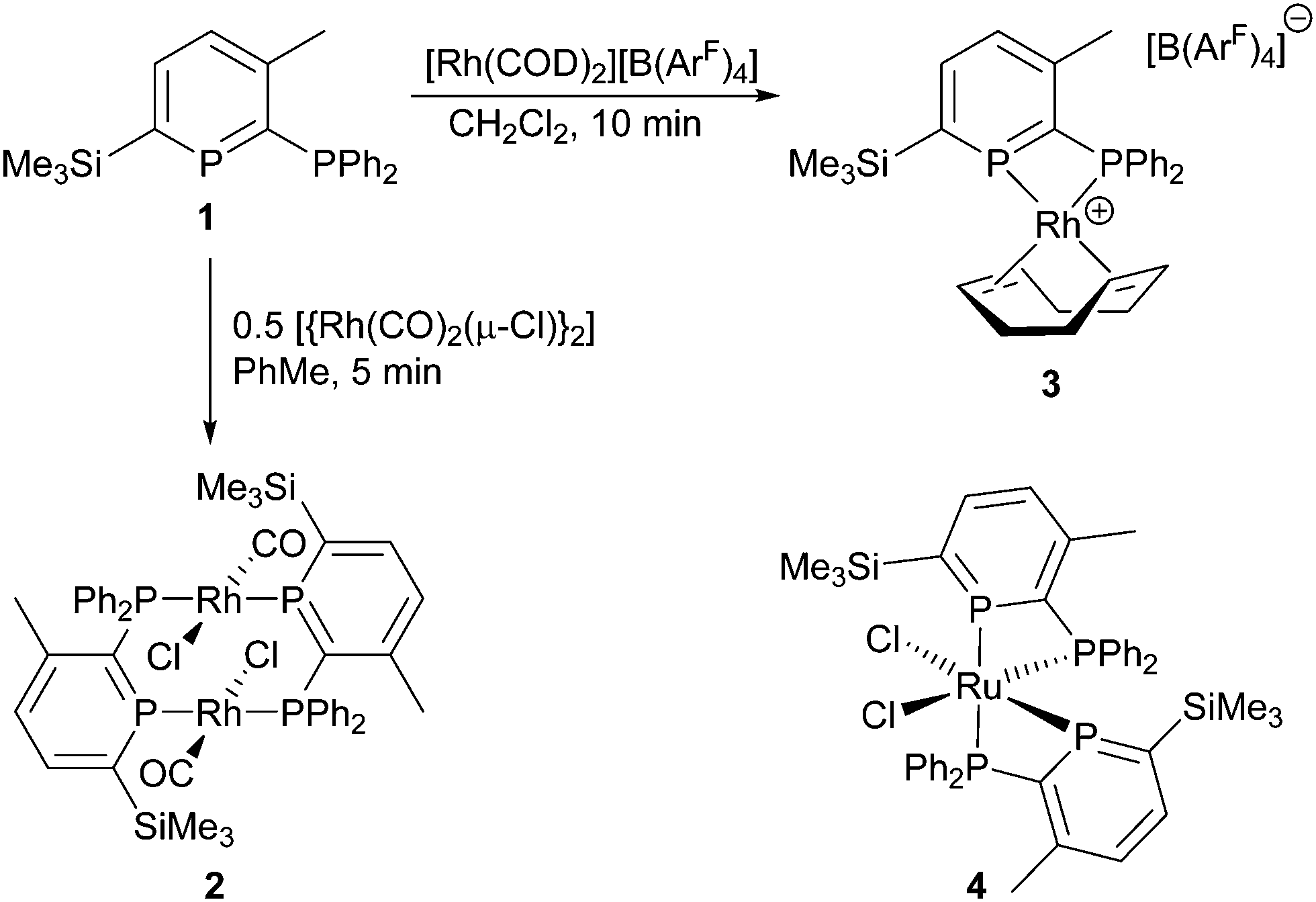 | ||
| Scheme 1 Synthesis of Rh complexes of 1, and the structure of 4. | ||
Initially, we probed the coordination properties of 1 using [{Rh(CO)2(μ-Cl)}2] (Scheme 1). A rapid reaction was observed affording a deep purple solution and the expected evolution of carbon monoxide. Crystalline 2 precipitated in 63% yield and was characterised as a bridged dinuclear complex by multinuclear NMR spectroscopy, high-resolution mass spectrometry, IR spectroscopy, X-ray diffraction and elemental analysis. 31P{1H}-NMR spectroscopy revealed the formation of a single product with two apparent doublet-of-triplets resonances (Fig. S2 and S3, ESI†) observed at δ = 250.6 ppm (phosphinine P) and 25.5 ppm (PPh2) that were successfully simulated (ESI†). Although a chelating complex was not achieved, 2 did facilitate comparisons to dppm and other diphosphines using the resulting carbonyl stretching frequency.26 FTIR spectroscopy revealed a single carbonyl stretch at ν = 1977 cm−1 which correlates with 1 being a more π-accepting ligand than dppm (ν = 1968 cm−1 for the analogous complex).26 The molecular structure of 2 (Fig. 1) displayed two bridging ligands with the CO and Cl ligands disordered across two positions. The Rh2(PCP)2 unit is non-planar, and has a dihedral angle for P(1)–Rh(1)–Rh(2)–P(2) of 16.88(4)°. Whilst stable in the solid state, 2 appears to be unstable in solution after prolonged periods or when heated (Fig. S11, ESI†).
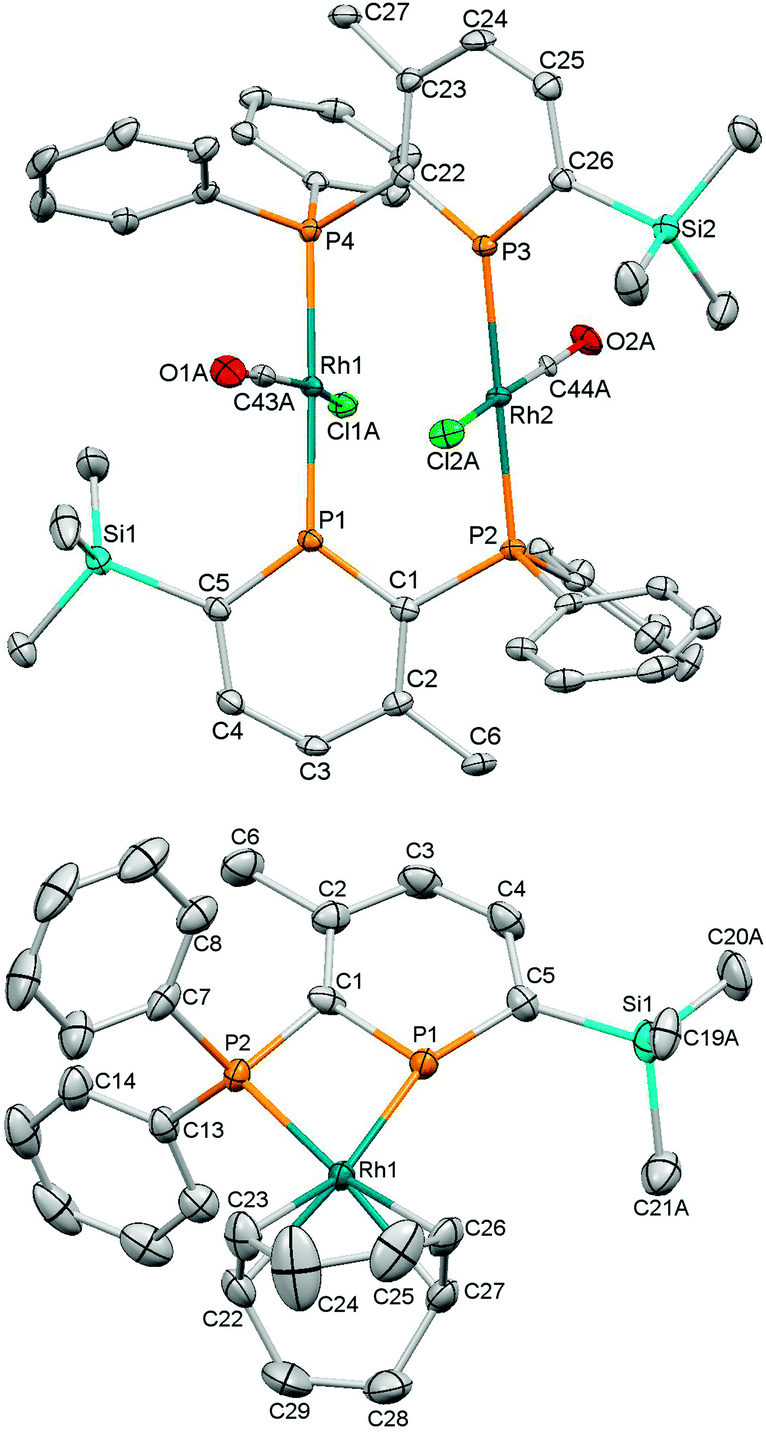 | ||
| Fig. 1 Thermal ellipsoid plots (50%) of the molecular structures of 2 (top) and 3 (bottom, B(ArF)4 anion excluded for clarity). Selected bond distances (Å) and angles (°) for 2: P(1)–Rh(1) 2.2845(9), P(2)–Rh(2) 2.3228(10), P(1)–C(1) 1.734(4), C(1)–C(2) 1.411(5), C(2)–C(3) 1.402(5), C(3)–C(4) 1.386(5), C(4)–C(5) 1.394(5), C(5)–P(1) 1.725(3), C(1)–P(2) 1.844(3), C(1)–P(1)–C(5) 107.6(2), P(1)–C(1)–P(2) 114.9(2); For 3: P(1)–Rh(1) 2.2932(8), P(2)–Rh(1) 2.2941(7), P(1)–C(1) 1.732(3), C(1)–C(2) 1.397(4), C(2)–C(3) 1.397(5), C(3)–C(4) 1.385(5), C(4)–C(5) 1.409(4), C(5)–P(1) 1.723(3), C(1)–P(2) 1.801(3), C(1)–P(1)–C(5) 106.9(2), P(1)–C(1)–P(2) 97.3(1), P(1)–Rh(1)–P(2) 70.64(3). | ||
Monodentate phosphinine ligands have been structurally characterised binding to Rh in trans-[Rh(CO)(L)2Cl],21d [Rh(L)2(COD)]+,27 and homoleptic [Rh(L)4]+ complexes (see ESI†).21c Rh complexes with tri-28 and tetraphosphinine29 ligands have also been structurally characterised (see ESI†).
In order to develop a mononuclear complex with a bidentate phosphinophosphinine ligand, a chelating co-ligand was utilised. Typically, sterically bulky ligands, such as dcpm (bis(dicyclohexylphosphino)methane),30 are required to stabilise small bite-angle cationic [Rh(diphosphine)(COD)]+ complexes31 due to the Thorpe–Ingold effect,22 and no chelating [Rh(dppm)(COD)]+ complexes have been structurally characterised. Our initial efforts focused on reaction of 1 with [{Rh(COD)(μ-Cl)}2] in the presence of a silver salt (AgBF4, AgSbF6) in CH2Cl2.32 However, we observed multiple products, even with slow addition of 1 using dilute conditions. We were inspired by the use of a bis(cyclooctadiene) complex of Rh using the weakly coordinating anion B(ArF)4 (tetrakis[3,5-bis(trifluoromethyl)phenyl]borate),33 and observed a rapid reaction of this precursor with 1, forming a single air-stable product in an 80% yield (Scheme 1). Complex 3 was characterised by X-ray diffraction, multinuclear NMR spectroscopy, high-resolution mass spectrometry and elemental analysis. 31P{1H}-NMR spectroscopy revealed two sets of apparent doublets-of-doublets at δ = 189.4 ppm and −6.8 ppm. The clean formation of 3 and its stability is noteworthy with such an acute P–Rh–P bite angle of 70.64(3)° despite the minimal steric bulk on both donors.§ With two complexes in hand, and as there were no previous reports of Rh-catalysts for carbonyl hydroboration in the literature, a catalyst screen of Rh complexes and common phosphine ligands was conducted using 4′-bromoacetophenone and catecholborane (Table 1).
|
|
||
|---|---|---|
| Run | Catalyst | Yieldc,d (%) |
| a Premixed in THF (∼0.1 ml) for 10 min before reaction. b Premixed in C6D6 (∼0.1 ml) for 10 min before reaction. c Yield measured against 1,3,5-trimethoxybenzene internal standard. d Complete 1H-NMR data for all catalytic runs available (ESI). Cat = catecholate: 1,2-(O)2C6H4. | ||
| A | — | 1 |
| B | 1 | 20 |
| C | 2 | 35 |
| D | 3 | 96 |
| E | 4 | 12 |
| F | [Rh(PPh3)3Cl] | 3 |
| G | [Rh(COD)2][B(ArF)4] + 2 PCy3a | 22 |
| H | 0.5 [{Rh(COD)Cl}2] + 2 PCy3b | 18 |
| I | [Rh(COD)2][B(ArF)4] + 2 PPh3a | 7 |
| J | [Rh(COD)2][B(ArF)4] + dppma | 7 |
| K | [Rh(COD)2][B(ArF)4] + 2 P(OPh)3a | 6 |
An initial test using 0.1 mol% 3 (run D) gave rapid conversion to the boronate ester, with a 94% yield observed within ten minutes and the reaction essentially complete after 30 minutes. A dramatic decrease in yield was observed when either the free ligand 1 (run B)¶ or complexes 2 and 4 were used (runs C and E), although 2 still proved to be more active than other Rh precursor/ligand combinations that were tested, including Wilkinson's catalyst (run F), which is a standard catalyst for alkene hydroboration.17b,f Only mixtures of [Rh(COD)2] [B(ArF)4] or [{Rh(COD)Cl}2] and PCy3 (tricyclohexylphosphine, runs G and H respectively) gave higher than a 10% yield. Tests using [Rh(COD)2][B(ArF)4] with less σ-donating PPh3 and dppm ligands in a 1:2 or 1:1 ratio respectively (runs I and J) gave similar, low yields. Finally, to test if the π-accepting properties of 1 were the source of the increased activity of 3, P(OPh)3 (run K) was tested, however, a similar low yield was obtained.
Having established the catalytic activity of 3, a screen of readily available acetophenone derivatives was carried out (Table 2). With the substrates tested, clean formation of the desired boronate ester was observed, with the exception of 4′-methoxyacetophenone, which produced multiple unidentified products (see Fig. S51, ESI†). Complex 3 also acted as a catalyst for the hydroboration of benzaldehyde, although the uncatalysed reaction also proceeds readily. Ketimines were then tested as more challenging substrates, and although room temperature reactions proceeded slowly, heating to 50 °C achieved acceptable yields within 1 hour. However, heating the reaction further gave no observable increase in yield. In contrast to the carbonyl substrates, installation of an electron-donating or withdrawing substituent on the C–Ar ring made little difference to the obtained yield, whereas the presence of a nitro-group on the N–Ar substituent severely hindered the reaction.
|
|
||||||
|---|---|---|---|---|---|---|
| E | R1 | R2 | R3 | Yieldd (%) | ||
| 10 min | 30 min | 60 min | ||||
| Conditions: 3, substrate (0.43 mmol), catecholborane (0.47 mmol, 1.1 eq.), C6D6 (0.6 cm3).a 0.1 mol% 3, 25 °C.b No catalyst, 25 °C.c 1 mol% 3, 50 °C.d Yields measured against 1,3,5-trimethoxybenzene internal standard. Cat = 1,2-(O)2C6H4. | ||||||
| Oa | CH3 | H | H | 91 | 97 | — |
| Oa | CH3 | H | Br | 94 | 96 | — |
| Oa | CH3 | H | F | 97 | >99 | — |
| Oa | CH3 | H | NO2 | 95 | 98 | — |
| Oa | CH3 | H | CH3 | 66 | 90 | 92 |
| Oa | CH3 | OCH3 | H | 97 | 99 | — |
| Ob | H | H | H | 52 | 75 | 86 |
| Oa | H | H | H | 95 | >99 | — |
| N-Phc | CH3 | H | H | — | — | 86 |
| N-Phc | CH3 | H | F | — | — | 86 |
| N-Phc | CH3 | H | CH3 | — | — | 85 |
| N-(p-NO2-C6H4)c | CH3 | H | H | — | — | 16 |
Complex 3 was also a competent catalyst for the hydroboration of the N-heterocycles acridine and quinoline, and was shown to be active in the catalytic hydrogenation of styrene and cyclohexene (see ESI† for details).
In conclusion, we have synthesised and characterised the first two Rh complexes of a 2-phosphinophosphinine. Both were tested in the catalytic hydroboration of 4′-bromoacetophenone as well as a previously reported ruthenium phosphinophosphinine complex, Wilkinson's catalyst and a series of commonly used phosphine ligands. The results clearly demonstrated that significant activity is only observed for the chelating complex 3, with high catalytic activity observed for several acetophenone derivatives at 0.1 mol% catalytic loading. Hydroboration of the more challenging N-phenyl ketimine substrates was also achieved, with good conversion in 1 hour at 1 mol% loading. Control reactions showed that simple electronic or bite-angle effects, as shown by the poor activity of different conventional monophosphine ligands and dppm, do not explain this catalytic activity. Future work will look to identify whether metal–ligand cooperativity or the hybrid nature of the ligand is playing a key role in generating highly active Rh catalysts.
The authors would like to acknowledge the EPSRC UK National Mass Spectrometry Facility at Swansea University, and Fluorochem for providing samples of the N-heterocycles. Dr Gary Nichol (University of Edinburgh) is acknowledged for collecting X-ray diffraction data for 3. Financial support is gratefully acknowledged from the EPSRC (DTP studentship to RJN), the Royal Society (Research grant: RG130436) and Heriot-Watt University as well as the UK Catalysis Hub Consortium (funded by EPSRC grants EP/K014706/2, EP/K014668/1, EP/K014854/1, EP/K014714/1 and EP/M013219/1) for providing travel funding to SMM and RJN.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- C. C. Chong and R. Kinjo, ACS Catal., 2015, 5, 3238 CrossRef
.
- J. Magano and J. R. Dunetz, Org. Process Res. Dev., 2012, 16, 1156 CrossRef
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621 CrossRef PubMed
- R. McLellan, A. R. Kennedy, R. E. Mulvey, S. A. Orr and S. D. Robertson, Chem. – Eur. J., 2017, 23, 16853 CrossRef PubMed
- Y. Wu, C. Shan, J. Ying, J. Su, J. Zhu, L. L. Liu and Y. Zhao, Green Chem., 2017, 19, 4169 RSC
- M. Arrowsmith, T. J. Hadlington, M. S. Hill and G. G. Kociok-Köhn, Chem. Commun., 2012, 48, 4567 RSC
- J. R. Lawson, L. C. Wilkins and R. L. Melen, Chem. – Eur. J., 2017, 23, 10997 CrossRef PubMed
-
(a) V. K. Jakhar, M. K. Barman and S. Nembenna, Org. Lett., 2016, 18, 4710 CrossRef PubMed
- T. J. Hadlington, M. Hermann, G. Frenking and C. Jones, J. Am. Chem. Soc., 2014, 136, 3028 CrossRef PubMed
- A. A. Oluyadi, S. Ma and C. N. Muhoro, Organometallics, 2013, 32, 70 CrossRef
- G. Zhang, H. Zeng, J. Wu, Z. Yin, S. Zheng and J. C. Fettinger, Angew. Chem., Int. Ed., 2016, 55, 14369 CrossRef PubMed
- S. R. Tamang and M. Findlater, J. Org. Chem., 2017, 82, 12857 CrossRef PubMed
- A. E. King, S. C. E. Stieber, N. J. Henson, S. A. Kozimor, B. L. Scott, N. C. Smythe, A. D. Sutton and J. C. Gordon, Eur. J. Inorg. Chem., 2016, 1635 CrossRef
- A. Y. Khalimon, P. Farha, L. G. Kuzmina and G. I. Nikonov, Chem. Commun., 2012, 48, 455 RSC
- R. Arevalo, C. M. Vogels, G. A. MacNeil, L. Riera, J. Perez and S. A. Westcott, Dalton Trans., 2017, 46, 7750 RSC
-
(a) L. Koren-Selfridge, H. N. Londino, J. K. Vellucci, B. J. Simmons, C. P. Casey and T. B. Clark, Organometallics, 2009, 28, 2085 CrossRef
-
(a) D. Männig and H. Nöth, Angew. Chem., Int. Ed. Engl., 1985, 24, 878 CrossRef
- M. W. Drover, L. L. Schafer and J. A. Love, Angew. Chem., Int. Ed. Engl., 2016, 55, 3181 CrossRef PubMed
-
(a)
C. Müller, Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, John Wiley & Sons, Ltd, 2012, pp. 287–307 Search PubMed
-
(a) L. E. E. Broeckx, A. Bucci, C. Zuccaccia, M. Lutz, A. Macchioni and C. Müller, Organometallics, 2015, 34, 2943 CrossRef
-
(a) B. Breit, J. Mol. Catal. A: Chem., 1999, 143, 143 CrossRef
- S. M. Mansell, Dalton Trans., 2017, 46, 15157 RSC
- P. L. Floch, Coord. Chem. Rev., 2006, 250, 627 CrossRef
- R. J. Newland, M. F. Wyatt, R. L. Wingad and S. M. Mansell, Dalton Trans., 2017, 46, 6172 RSC
- R. J. Newland, A. Smith, D. M. Smith, N. Fey, M. J. Hanton and S. M. Mansell, Organometallics, 2018, 37, 1062 CrossRef
- A. R. Sanger, J. Chem. Soc., Chem. Commun., 1975, 893 RSC
- M. Doux, L. Ricard, F. Mathey, P. L. Floch and N. Mézailles, Eur. J. Inorg. Chem., 2003, 687 CrossRef
- N. Mézailles, N. Avarvari, L. Ricard, F. Mathey and P. Le Floch, Inorg. Chem., 1998, 37, 5313 CrossRef
- N. Avarvari, N. Mézailles, L. Ricard, P. L. Floch and F. Mathey, Science, 1998, 280, 1587 CrossRef PubMed
- A. L. Colebatch, A. I. McKay, N. A. Beattie, S. A. Macgregor and A. S. Weller, Eur. J. Inorg. Chem., 2017, 4533 CrossRef
-
(a) I. D. Gridnev, Y. Liu and T. Imamoto, ACS Catal., 2014, 4, 203 CrossRef
- S. A. Bhat, M. K. Pandey, J. T. Mague and M. S. Balakrishna, Dalton Trans., 2017, 46, 227 RSC
- A. B. Chaplin, J. F. Hooper, A. S. Weller and M. C. Willis, J. Am. Chem. Soc., 2012, 134, 4885 CrossRef PubMed
- I. Beletskaya and A. Pelter, Tetrahedron, 1997, 53, 4957 CrossRef
- D. A. Evans and A. H. Hoveyda, J. Org. Chem., 1990, 55, 5190 CrossRef
- C. M. Vogels, P. E. O'Connor, T. E. Phillips, K. J. Watson, M. P. Shaver, P. G. Hayes and S. A. Westcott, Can. J. Chem., 2001, 79, 1898 CrossRef
- R. T. Baker, J. C. Calabrese and S. A. Westcott, J. Organomet. Chem., 1995, 498, 109 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available: Additional synthetic details, analytical data and crystallographic data in CIF format. Additional research data supporting this publication are available from Heriot-Watt University's research data repository at DOI: http://10.17861/14c23fe6-bc65-4806-ba5e-63642a6ad3e9. CCDC 1822415 and 1822416. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc01880d |
| ‡ No catalysts for the hydroboration of ketones were mentioned in several key reviews1,17g,34 or in additional thorough searches of the literature. Results by Männig & Nöth indicate that hydroboration of aliphatic ketones proceeds selectively over alkene hydroboration without a catalyst,17a however, this is not the case for aryl ketones at 25 °C (run A, Table 1). Evans & Hoveyda demonstrated that hydroboration of β-hydroxyketones in the presence of catalytic amounts (5 mol%) of Wilkinson's catalyst provides some measure of increased diastereocontrol, however, no improvements in reaction rate or conversion were observed.35 Westcott et al. demonstrated the Rh catalysed hydroboration of aldimines with HBCat,36 but it was previously reported that bulkier N-phenyl aldimines can react rapidly with HBCat without the need for a catalyst.37 |
| § For a histogram of Rh(κ2-PEP) bite angles of entries found in the CSD (E = C, N, O), see Fig. S24 (ESI†). |
| ¶ Thus confirming that the high yield observed for 3 was not a result of the ligand dissociating during the catalytic run. With 0.1 mol% 1, the reaction gave a 95% yield after 14 hours. Upon mixing 1 and HBCat in C6D6, no Lewis adduct was observed. |
| This journal is © The Royal Society of Chemistry 2018 |