
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Silicon-fluorine chemistry: from the preparation of SiF2 to C–F bond activation using silylenes and its heavier congeners†
Sakya S.
Sen
 *a and
Herbert W.
Roesky
*b
*a and
Herbert W.
Roesky
*b
aInorganic Chemistry and Catalysis Division, CSIR-National Chemical Laboratory, Dr Homi Bhabha Road, Pashan, Pune 411008, India
bInstitute of Inorganic Chemistry, Georg-August University, Tammannstrasse 4, D-37077, Goettingen, Germany. E-mail: hroesky@gwdg.de
First published on 10th April 2018
Abstract
This feature article is intended to provide a background to the history of the isolation of silicon(II) fluorides and the different synthetic methodologies used to generate them. Although first detected in the 1970s, the chemistry of silicon(II) fluorides has not encountered serious research efforts for a rather long period of time. This is somewhat surprising given the fact that the chemistry of compounds with divalent silicon has undergone a renaissance during last three decades. Recently, the interest in silicon(II) fluorides have been resparked with tremendous progress being achieved in this area, in particular, with respect to their synthesis and structural characterisation. The successful isolation of cyclic alkyl amino carbene (cAAC) stabilized silicon difluoride has completed the classic progression of SiF2, from a transient intermediate to spectroscopically detected molecule to a stable compound. The related germanium(II), tin(II), and lead(II) fluoride chemistry will also be discussed. Apart from the isolation of tetrel(II) fluorides, the use of compounds with low valent group 14 elements for the selective activation and functionalisation of C–F bonds has witnessed some remarkable advances, which will also be summarized in this feature article.
 Sakya S. Sen | Sakya S. Sen received his PhD from the University of Göttingen, Germany under the supervision of Professor Herbert W. Roesky and subsequently joined the laboratory of Professor Holger Braunschweig at the University of Würzburg, Germany as an AvH postdoctoral fellow. Since 2014 he has worked in the CSIR-NCL, Pune, India as a senior scientist. His research interests are the synthesis and application of compounds with low valent main group elements. Sakya is the recipient of the CSIR-young scientist award in 2017 and young associate of Indian Academy of Sciences. |
 Herbert W. Roesky | Herbert W. Roesky obtained his doctorate from the University of Göttingen. After working at DuPont in the United States, he returned to Göttingen and finished his habilitation. He then became a Professor at the Johann-Wolfgang-Goethe-Universität, Frankfurt am Main in 1971. He moved to the University of Göttingen in 1980 and was the director of the Institute for Inorganic Chemistry until 2004. He is primarily known for his pioneering work on fluorides of both transition and main group elements. His current research interest is focused on the synthesis and reactivity of compounds with heavier group 13 and 14 elements in low oxidation states. More than 1250 peer-reviewed papers, articles, patents, and books record his research activities in the areas of inorganic chemistry and materials science. He is also the recipient of several prizes, i.e. Gottfried Wilhelm Leibniz Prize, the Alfred Stock Memorial Prize, the Grand Prix de la Foundation de la Maison de la Chimie, the Wilkinson Prize, Moissan award and ACS awards in Inorganic and Fluorine Chemistry. |
Introduction
“Oh threats of hell and hopes of paradise!One thing at least is certain – This Life flies”
– Omar Khayyám, Rubaiyat of Omar Khayyam
The austere insight of Khayyám about life bears a striking resemblance to the present day silicon chemistry. Despite the ever-looming threat of instability and high reactivity of organosilicon compounds, especially in the low oxidation state, and the hope of taming them using chemical tricks (bulky ligands, donor–acceptor approach), the only certain thing is that the present day silicon chemistry is propelling in fifth gear. Starting from the seminal report on the synthesis of a compound with a Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si double bond by West and co-workers1 that toppled the then much venerated “double bond rule”, to a stable silanone,2 which was “F. S. Kipping's dream”, organosilicon chemistry has traversed a long journey, witnessing a catalogue of esteemed discoveries made across the world.3 Such realisation of novel organosilicon compounds has allowed us to analyze their structures, elucidate their bonding properties and study their reactivities, which eventually revealed the marked differences between silicon and carbon, and led W. Kutzelnigg to comment “the heavier main-group elements, Si to Pb, actually exhibit ‘normal’ chemical behaviour while carbon should be considered the ‘unusual’ member of the group 14 elements rather than the prototype”.4 The remarkable progress of organosilicon chemistry over the last three decades has been closely monitored by the chemistry community, which has resulted in numerous elegant reviews.5
Si double bond by West and co-workers1 that toppled the then much venerated “double bond rule”, to a stable silanone,2 which was “F. S. Kipping's dream”, organosilicon chemistry has traversed a long journey, witnessing a catalogue of esteemed discoveries made across the world.3 Such realisation of novel organosilicon compounds has allowed us to analyze their structures, elucidate their bonding properties and study their reactivities, which eventually revealed the marked differences between silicon and carbon, and led W. Kutzelnigg to comment “the heavier main-group elements, Si to Pb, actually exhibit ‘normal’ chemical behaviour while carbon should be considered the ‘unusual’ member of the group 14 elements rather than the prototype”.4 The remarkable progress of organosilicon chemistry over the last three decades has been closely monitored by the chemistry community, which has resulted in numerous elegant reviews.5
Even before the beginning of the chemistry of compounds with multiple-bonds between silicon atoms, the chemistry of silicon(II) halides has attracted considerable attention. The confirmation of the: CCl2 intermediate in the Reimer–Tiemann reaction,6a as well as Nef's pioneering work6b,c on divalent carbon compounds have led silicon chemists to undertake the synthesis of dihalosilylenes. Another impetus originated from the realisation that silicon(II) halides are the intermediates in many important reactions, such as the Rochow Müller process,7a where methylchlorosilanes are prepared from methyl chloride in an easy and inexpensive way. The Rochow Müller process has resulted in the huge growth of the silicone polymer area. Spectral evidence has been obtained in the formation of unstable silylenes (SiCl2 and MeSiCl) during the reaction. Such silicon(II) compounds can be used as synthons to prepare new compounds, which are otherwise inaccessible. In their recent feature article on the readily available SiCl2/[SiCl3]− system, Teichmann and Wagner elegantly highlighted the classical high-temperature protocols used for the generation of SiCl2 and their utility as synthons for making unusual silicon compounds.7b
Following the syntheses of [SiBr2]x and [SiCl2]x by the Schmeißer group,8 chemists have turned their attention towards [SiF2]x. The interest has been stimulated by the fact that the divalent fluoride of carbon is more stable than its parent carbene. For example, carbene (:CH2) has a half-life of 1 ms in the gas phase, while carbon difluoride has a half-life of 1 s.9a,b The half-life of gaseous SiF2 is 150 s at 0.2 Torr and ambient temperature.9c Moreover, neither CF4 nor SiF4 react with glass, which is a usual problem for other fluorides. The preparation of SiF2 from magnesium and dibromofluorosilane by Schmeißer goes back as early as 1954.10 Since then, the divalent fluorides of silicon have become a major research area especially at Rice University, USA where the Timms and Margrave groups have performed an extensive and systematic study of the properties of silicon difluoride.11 Nevertheless, no monomeric SiF2 has been isolated under synthetically useful conditions until very recently, when we reported cyclic alkyl amino carbene (cAAC) coordinated silicon difluoride from the reduction of cAAC·SiF4 with KC8.12 In this review, we provide a comprehensive coverage of the synthesis and reactions of silicon(II) fluorides. The fluorides of other heavier group 14 elements such Ge(II), Sn(II) and Pb(II) will be covered. It will also summarize the recent efforts, by our group and others, on the activation of the C–F bond using compounds with low valent silicon atoms. We extend into other classes of reactivity that are closely related and of particular interest such as germylene and stannylene assisted C–F activation. Finally, the recent studies on the hydrodefluorination of C−F bonds using silylium cations are also presented.
Early preparations of silicon difluoride
Table 1 lists some key milestones in the development of group 14-fluoride chemistry. Although the groups of Timms and Margrave at Rice University, Houston, USA are generally credited for preparing SiF2 for the first time, Schmeißer prepared polymeric silicon(II) fluoride from the reaction of dibromofluorosilane and magnesium in 1954.10 In 1958, Pease found that silicon difluoride could be formed from the reaction of SiF4 and Si at high temperature, which condenses below −80 °C.13 Another effort from Schmeißer and Ehlers involved the cleavage of the Si–Si bond in Si2F6 at 700 °C that led to the formation of SiF4 along with SiF2,14 which condensed as a yellow solid in liquid air. Above −80 °C, SiF2 polymerizes to yield a colorless, rubbery solid [SiF2]x, which catches fire in moist air.| Year | Comment |
|---|---|
| 1964 | Schmeißer and Ehlers observed the formation of SiF2 at −80 °C |
| 1965 | Timms et al. prepared SiF2 from SiF4, measured its half-life, and studied its reactivity |
| 2001 | Roesky and co-workers structurally characterized Ge(II) fluoride |
| 2008 | Roesky and co-workers structurally characterized Sn(II) fluoride |
| 2005 | Ozerov and co-workers used silylium cation for the hydrodefluorination reaction |
| 2010 | Compounds with low valent silicon atom can activate C–F bonds |
| 2011/2012 | By coordinating the lone pair of silylene to BH3 or a transition metal fragment, the first compound featuring an Si(II)–F bond was realized |
| 2018 | Using cAAC carbene, the SiF2 moiety is stabilized and structurally validated |
All the aforementioned reactions in fact deal with polymeric silicon difluoride but indicate the formation of monomeric SiF2 as an intermediate. Moreover, no physical properties of SiF2 were measured. To unequivocally prove the existence of monomeric SiF2, Timms et al. constructed a special apparatus (Fig. 1). SiF4 was passed into a reservoir and subsequently, using the needle valves, passed over chunks of heated silicon at 1200 °C, which afforded 50% SiF2.15 To avoid the disproportionation reaction, as soon as the SiF2 is formed, it is swept through the vacuum line to a point where it's physical properties or chemical reactivities could be measured.
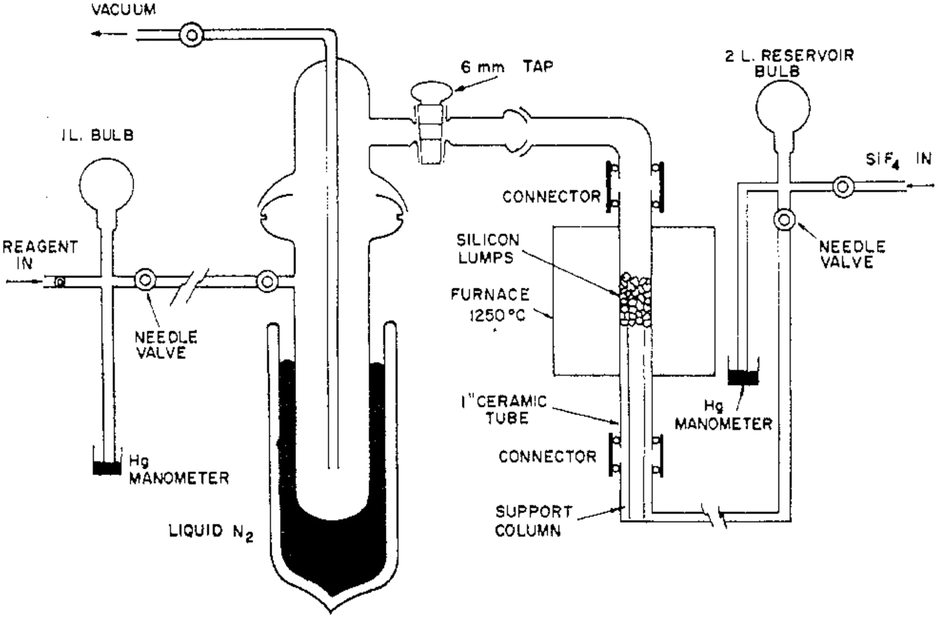 | ||
| Fig. 1 The apparatus used for the preparation and reaction of SiF2. Reproduced with permission from ref. 17. | ||
Ehlert and Margrave performed a mass spectrometric experiment of SiF2.16a SiF4 was passed through a Si column at very high temperature and subsequently passed into a mass spectrometer, which detected only SiF4 and monomeric SiF2; no polymeric species of SiF2 were observed. It was estimated that SiF2 has a half-life of 120 seconds. The infrared spectrum of gaseous SiF2 has been recorded from 1050 to 400 cm−1. Two absorption bands centered at 855 and 872 cm−1 were assigned to the symmetric and anti-symmetric stretching modes, respectively.16b
The relatively easy generation of SiF2 by the Timms’ group saw a flurry of research activity into their properties and reactivity in the early 1970s. The reactivity of polymeric SiF2 was investigated with various substrates such as BF3, benzene, hexafluorobenzene, and water (Scheme 1).17 The general strategy of the reactions was to condense the reactant and SiF4–SiF2 mixture together and characterize the condensate. Multi-nuclear NMR spectroscopy, IR spectroscopy, mass spectrometry, and elemental analysis were performed to characterize the condensates. As none of the product was characterized by single crystal X-ray diffraction, the structural authentication of the SiF2 unit remained elusive. In the following years, publications on silicon difluoride trickled out and there were not many significant advances. In fact, the research interest in silicon difluoride never again reached the heights that were attained in the 1960s and 70s.
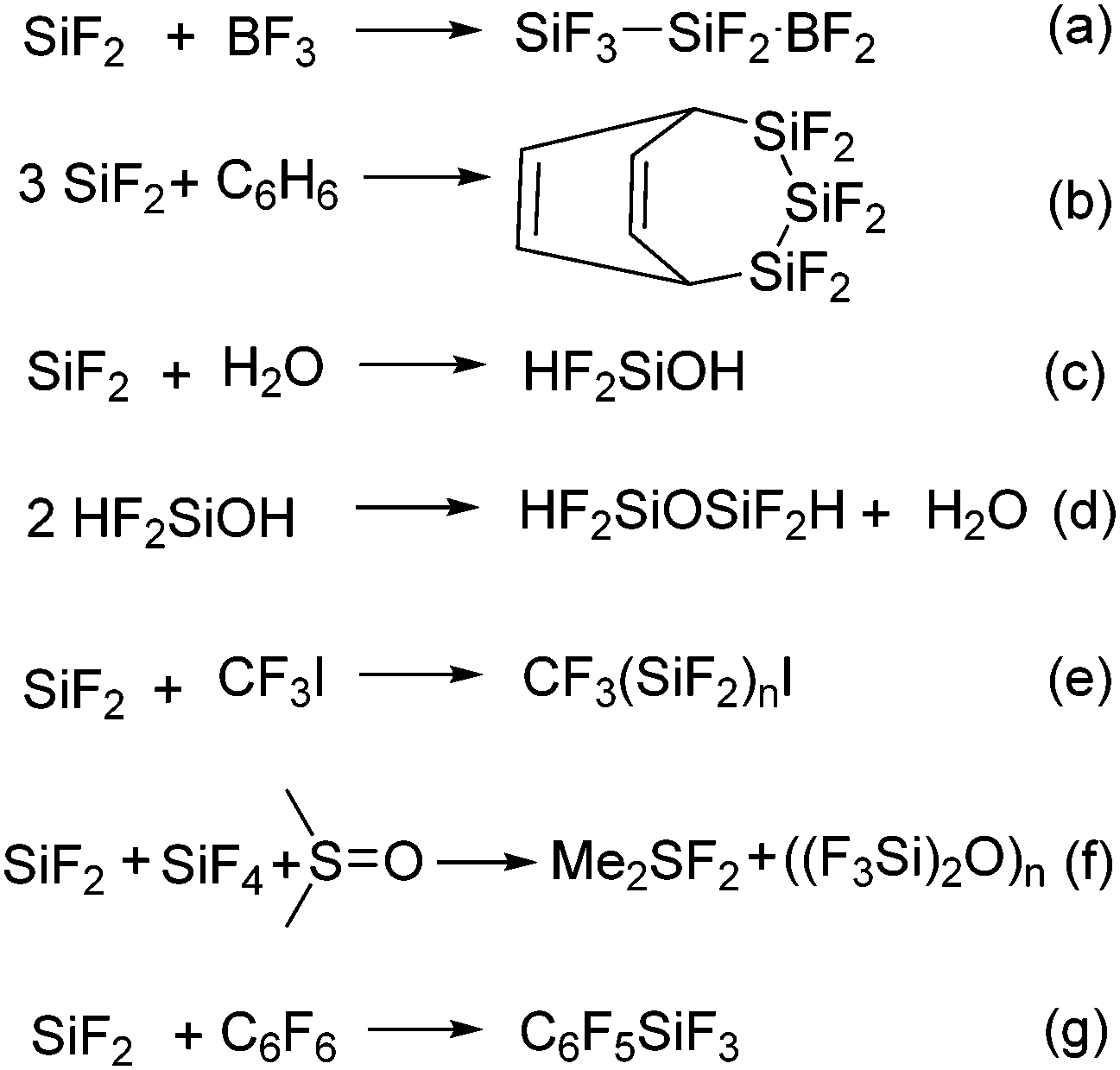 | ||
| Scheme 1 The reactions of a SiF4–SiF2 mixture with various substrates. | ||
The efforts to prepare stable compounds with an Si(II)–F bond: structural validation
The facile isolation of stable silicon(II) chlorides18 such as [LSi(II)Cl, L = {PhC(NtBu)2}−] (1) and N-heterocyclic carbene (NHC)-stabilized dichlorosilylene (L′Si(II)Cl2, L′ = 1,3-bis(2,6-iPr2C6H3)imidazol-2-ylidene) (2) renewed the interest in making a compound with a Si(II)–F bond. However, due to the high propensity of silicon(II) fluoride toward polymerization or disproportionation, attempts to isolate “[PhC(NtBu)2]SiF” or “IDipp·SiF2” were not successful. In line with our observations, Jutzi and co-workers also found that Me5C5SiF was unstable and dimerized into the corresponding disilene, Me5C5(F)SiSi(F)C5Me5. The latter was also found to be unstable and underwent a [2+2]-cycloaddition to form cyclotetrasilane.19
“We must accept finite disappointment, but never lose infinite hope.”
– Martin Luther King, Jr.
We imagined that the presence of the stereoactive lone pair of electrons on the silicon atom of silicon difluoride impedes its isolation. To prevent this feisty nature of silicon(II) difluoride, we reacted 1 with BH3 to coordinate the lone pair of electrons of silicon to the electron deficient boron center (Scheme 2).20 The reaction led to the formation of an adduct, 3. The latter was subsequently reacted with L1PbF, [L1 = CH{(CMe)(2,6-iPr2C6H3N)}2] (4),20 which resulted in the formation of compound 5 along with the formation of L1PbCl (6). 5 features a formal Si(II)–F bond. The 19F NMR spectrum displayed a resonance at δ −121.59 ppm accompanied by 29Si satellite signals (J = 438.17 Hz). The 29Si NMR spectrum revealed a doublet of quartets (δ 23.63 ppm and J(29Si–11B) = 66.78 Hz), which underpinned the presence of Si–B and Si–F bonds. Although the spectroscopic data unambiguously affirmed the formation of a compound with a Si(II)–F bond, its structural characterization by X-ray diffraction was still missing.
 | ||
| Scheme 2 The synthesis of amidinato stabilized Si(II) fluoride in the coordination sphere of borane; Ar = 2,6-iPr2-C6H3. | ||
The unraveling of an efficient synthetic pathway to access formal Si(II) fluorides using a donor–acceptor approach furthered us to bring transition metals into play instead of borane. A very common strategy that has often been applied in organometallic chemistry for isolating reactive compounds is to bind those species to transition metals.21 Moreover, the presence of a transition metal in a compound usually allows the growth of better quality single crystals suitable for X-ray analysis. Silylenes are known to coordinate with transition metals. Prior to us Rivard and co-workers described the generation of stable M(II)H2 (M = Si, Ge, and Sn) and H2SiEH2 (E = Ge and Sn) under the coordination sphere of late transition metals.22 By applying the same synthetic protocol, we prepared three silicon(II) fluoride compounds, PhC(NtBu)2(F)Si·M(CO)5 {M = Cr (10), Mo (11), and W(12)},23 using a metathesis reaction of the corresponding chlorides (7–9) with Me3SnF, a versatile fluorinating agent (Scheme 3). 10 and 12 were structurally characterized. There is a small variation in the Si–M bond lengths in 10 (2.3398(4) Å) and 12 (2.4990(8) Å) when compared to the corresponding chloride analogues 7 (2.3458(7) Å) and 9 (2.5086(11) Å). The Si–F bond lengths in 10 and 12 are 1.6168(8) and 1.6245(14) Å, respectively. A comparison of the Si−F bond lengths in 10 and 12 with that in gaseous SiF2 (1.59 Å) indicates an increase in the bond length by only 0.03 Å.
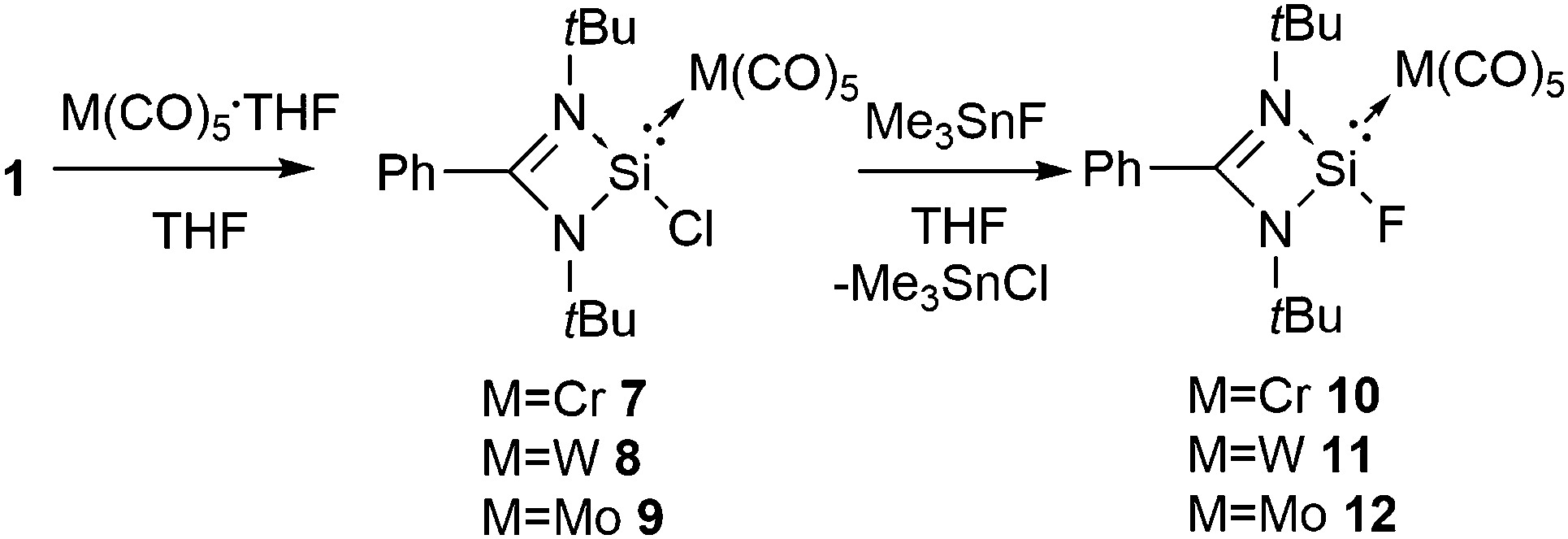 | ||
| Scheme 3 The synthesis of amidinato stabilized Si(II) fluorides in the coordination sphere of transition metals. | ||
“It's kind of fun to do the impossible.”
– Walt Disney
Although, 5 and 10–12 possess a formal Si(II)–F bond, access to a silicon difluoride that is stable at room temperature remained elusive. Therefore, the question arose whether it is viable to isolate a stable compound featuring a silicon(II) difluoride moiety. Since NHCs were found inappropriate for stabilizing the silicon(II) difluoride moiety, we turned our attention towards cAAC. According to the calculations,24 the singlet–triplet energy gap of cAAC (46 kcal mol−1) is much smaller than that of NHC (68 kcal mol−1), and as a result, the HOMO of cAAC (−5.0 eV) is higher in energy than that of NHC (−5.2 eV). Following the isolation of stable cAAC by the Bertrand group,25 it has been widely used for the stabilisation of a plethora of unprecedented silicon compounds that could be prepared in no other way.26 The attempts to synthesize cAAC supported SiF2 from the reactions of (cAAC)2SiCl2 with various fluorinating agents (Me3SnF, CsF, and C5F5N) were unsuccessful. Hence, a conventional reduction method was employed. The reduction of the cAAC–SiF4 adduct (13) with KC8 in the presence of another equivalent of cAAC resulted in (cAAC)2SiF2 (14) as a purple solid (Scheme 4).12 The 29Si and 19F NMR spectra of 14 display no resonance at room temperature due to their rapid exchange on the NMR timescale. However, a low temperature NMR experiment exhibited a triplet at δ 29.73 ppm in the 29Si NMR spectrum and a broad singlet at δ −123.47 ppm in the 19F NMR spectrum. The UV-vis spectrum of 14 showed an absorption band at 529 nm that was in good accordance with the TD-DFT calculations. This absorption can be attributed to ligand to silicon charge transfer. An X-ray crystal structure of 14 (Fig. 2) revealed that the SiF2 unit was coordinated by two cAAC moieties with Si–F bond lengths of 1.592(1) and 1.593(1) Å, which are within the range of those reported for 10. Subsequent theoretical studies showed that 14 possesses an open shell singlet configuration, which is 4.9 kcal mol−1 lower in energy than the triplet configuration and 10.2 kcal mol−1 lower in energy than the closed-shell singlet configuration. The large energy difference between the open shell singlet configuration and the triplet configuration underpins why 14 was EPR silent.
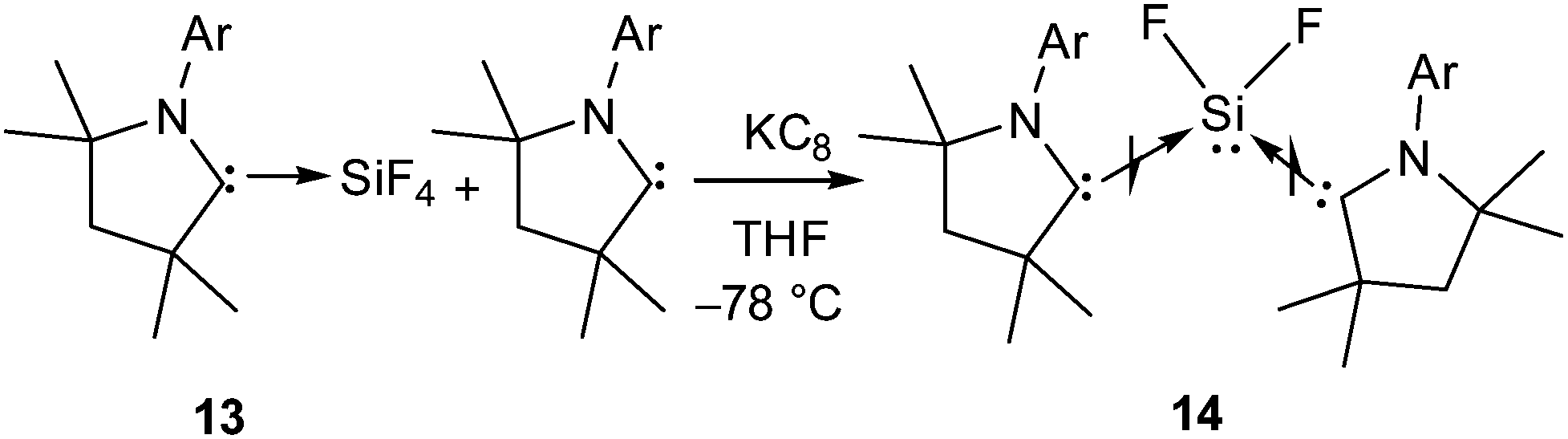 | ||
| Scheme 4 The synthesis of cAAC supported SiF2 (14) (Ar = 2,6-iPr2C6H3). | ||
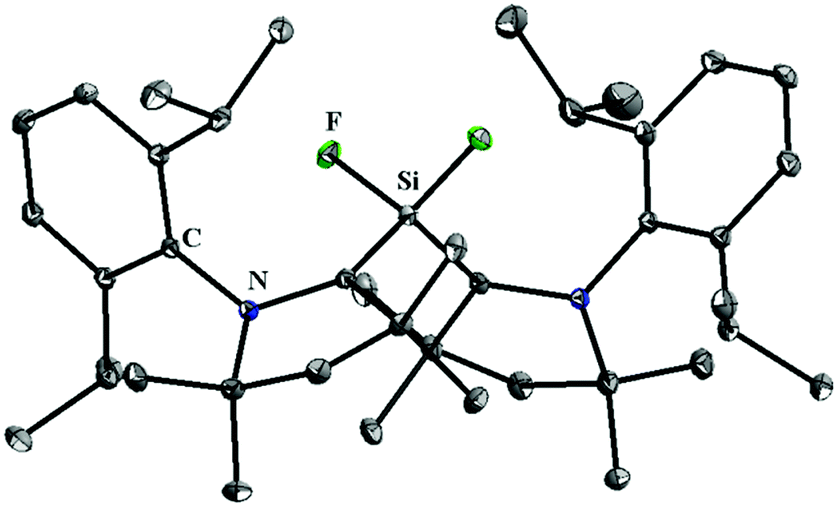 | ||
| Fig. 2 The molecular structure of 14. | ||
“There is no end. There is only the infinite passion of life”
–Federico Fellini (Italian Film Director)
Once we accomplished the isolation of a compound with a silicon(II) difluoride moiety, we wished to prepare a compound with a SiF3 radical unit. Gratifyingly, the reduction of 13 with one equivalent of KC8 in the absence of any free cAAC resulted in a monoradical product, (cAAC)SiF3 (15), which was the first stable radical containing a SiF3 group (Scheme 5).12 The monomeric nature of 15 was characterized using a single crystal X-ray diffraction study (Fig. 3). The EPR spectrum of 15 was recorded in n-hexane at room temperature (Fig. 4), which showed a very large 19F coupling parameter. According to the McConnell spin polarisation model, this high coupling parameter is due to the high polarity of the Si–F bond and its interaction with the carbon radical center.
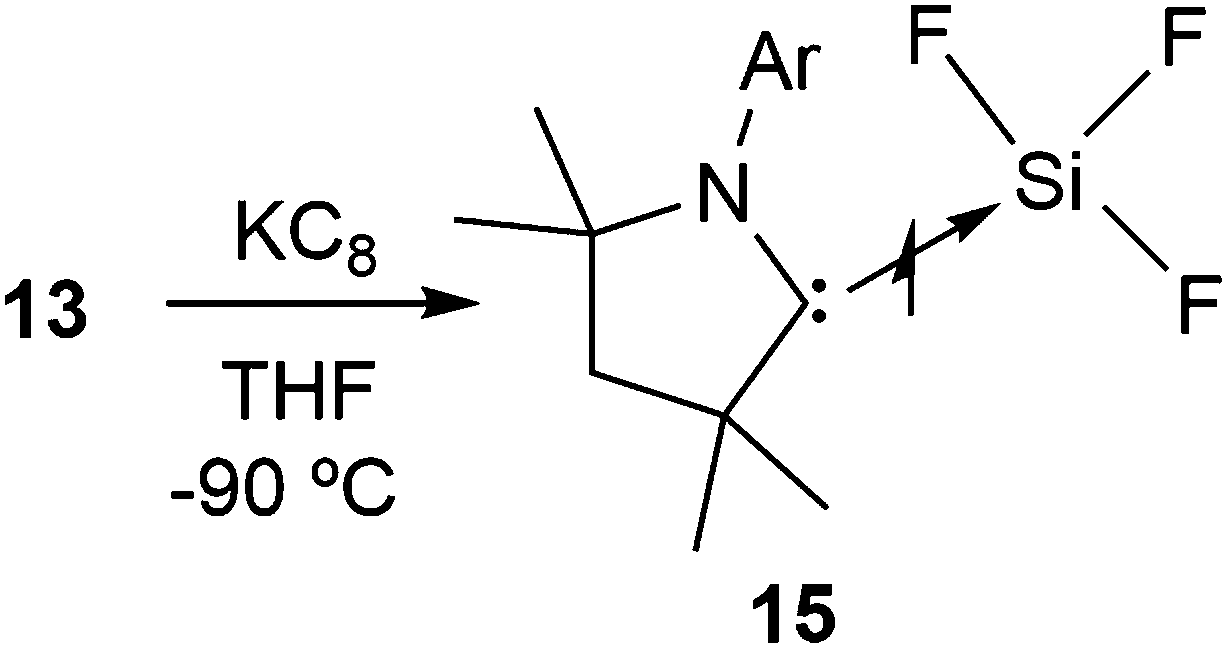 | ||
| Scheme 5 The synthesis of a cAAC supported SiF3 radical (15). | ||
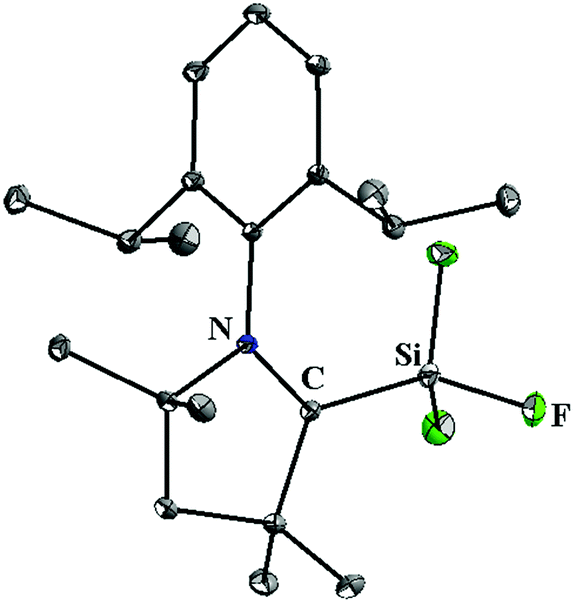 | ||
| Fig. 3 The molecular structure of 15. | ||
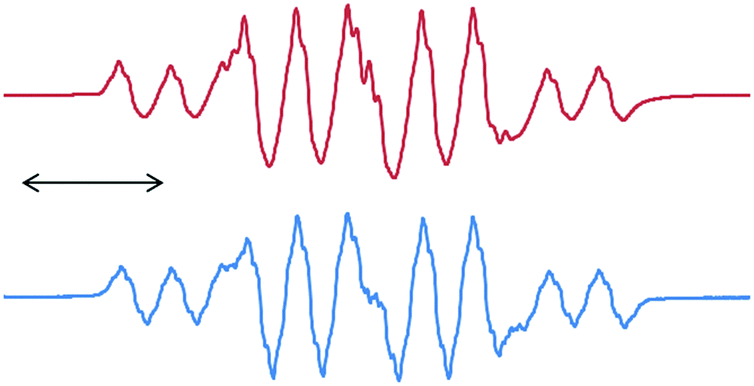 | ||
| Fig. 4 The experimental EPR spectrum of 15 (below) at room temperature recorded in hexane and the simulated spectrum (above) with a(14N) = 6.9 G, a(29Si) = 10 G, a(19F, 3F) = 16.8 G and a(1H, 3H) = 1.2 G. | ||
Germanium, tin and lead(II) fluorides
Due to the inert-pair effect, GeF2 and SnF2 are more stable than their lower homologues. GeF2 is a strongly fluorine-bridged chain polymer in which the parallel chains are cross-linked by weak fluorine bridges. SnF2 exists as a tetramer containing a puckered eight-membered ring in which the Sn and F atoms are alternately arranged. SnF2 has been widely used in toothpaste as a source of fluoride to harden the dental enamel. However, due to their polymeric structures GeF2 and SnF2 have low solubilities in common organic solvents and hence, it is difficult to study their reactions in the solution state.The preparation of compounds containing a Ge(II)–F bond was achieved with relative ease. Ge(II) fluorides, 18 and 19, were synthesized in our group from the reaction of Ge(II) monochlorides, L1GeCl (16) and L2GeCl [L2 = CH{(CMe)(2,6-Me2C6H3N)}2] (17) with Me3SnF in dichloromethane (Scheme 6).27 The 19F NMR spectra of 18 and 19 display a singlet resonance at δ 50.6 and 54.5 ppm, respectively. Single crystal X-ray diffraction studies showed that the Ge–F bond in 18 is 1.805(17) Å, which is in good accordance with the Ge–F bond in Inoue's [Ge(F)(NSIPr)2Ge][BF4] (SIPr = 1,3-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene) (1.800(4) Å).28
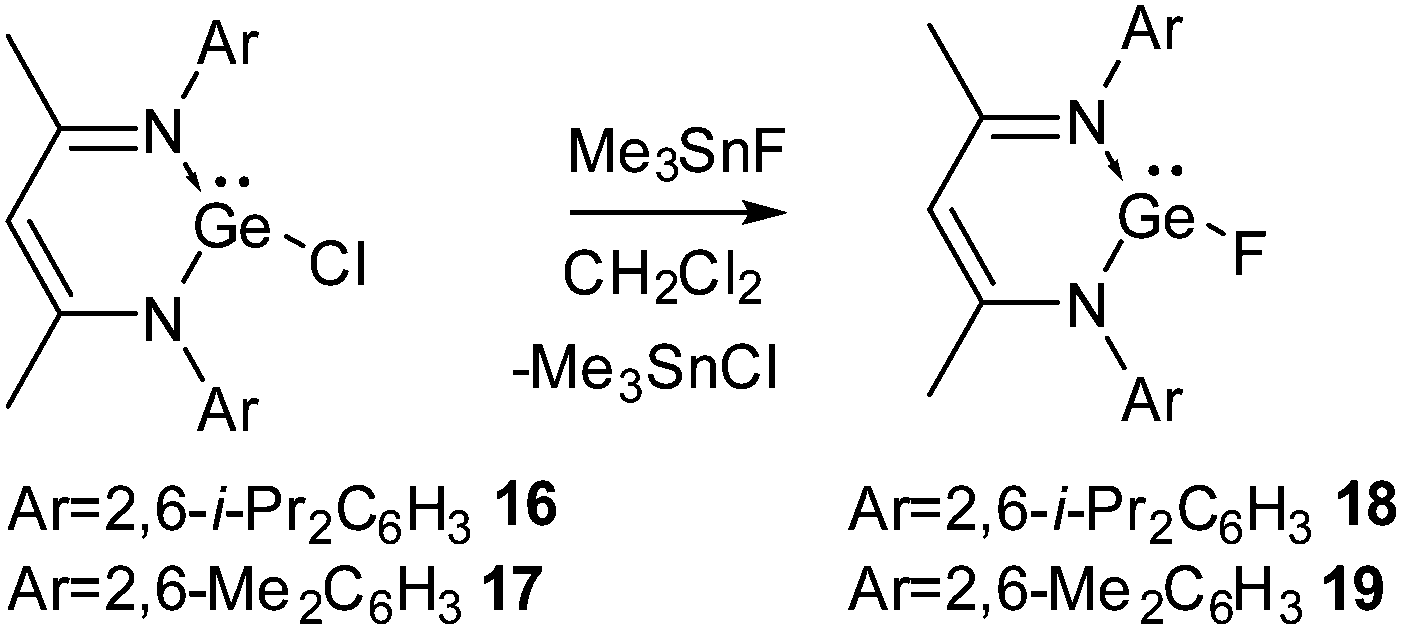 | ||
| Scheme 6 The synthesis of Ge(II) fluorides 18 and 19. | ||
The syntheses of 18 and 19 permitted us to investigate their oxidation reactions with organoazide and elemental sulfur. Refluxing a toluene solution of 18 and Me3SiN3 for 3 h resulted in the oxidative addition product 20 (Scheme 7).27 The oxidation of the Ge(II) center was accompanied by a downfield shift in the 19F NMR spectrum of 20 (δ 71.1 ppm). Treatment of 18 with elemental sulfur and selenium afforded {HC(CMeNAr)2}Ge(E)F (E = S (21)29a and Se (22),29b Ar = 2,6-iPr2C6H3) (Scheme 7). Alternatively, oxidation of 16 with elemental sulfur and selenium resulted in {HC(CMeNAr)2}Ge(E)Cl (E = S (26), Se (27)), which upon fluorination with Me3SnF furnished 21 and 22 (Scheme 8), respectively.29 The Ge–F bond length in 21 and 22 is 1.848(2) Å and 1.758(3) Å. The Ge–S bond distance in 21 is 2.050(9) Å, which is shorter than the reported Ge–S bond lengths in [{PhC(NtBu)2}2Ge2(μ-S)2Cl2] (2.2090(4) and 2.2978(4) Å)30 and the other reported Ge–S single bonds (2.17–2.25 Å)31 but comparable with those in 26 (2.050(9) Å) and germanethione, Tbt(Tipp)GeS (2.049(3) Å) [Tbt = 2,4,6-tris{bis(trimethylsilyl)methyl}phenyl and Tipp = 2,4,6-triisopropylphenyl].32 The Ge–Se bond length in 22 (2.176(7) Å) is very close to those in Tbt(Tipp)GeSe (2.180(2) Å)33 reported by Okazaki et al. and [(iBu)2ATI]Ge(Se)Cl (2.198(1) Å) (ATI = aminotroponiminate) reported by Nagendran and co-workers,34 but substantially shorter than the reported Ge–Se single bond lengths (2.337–2.421 Å).33,35 Treatment of alkyl lithium (Ar = Me, nBu) with 21 and 22 resulted in the dehalogenated derivatives [{HC(CMeNAr)2}Ge(E)R] (E = S, R = Me (23); E = Se, R = Me (24), nBu (25)) (Scheme 7).29
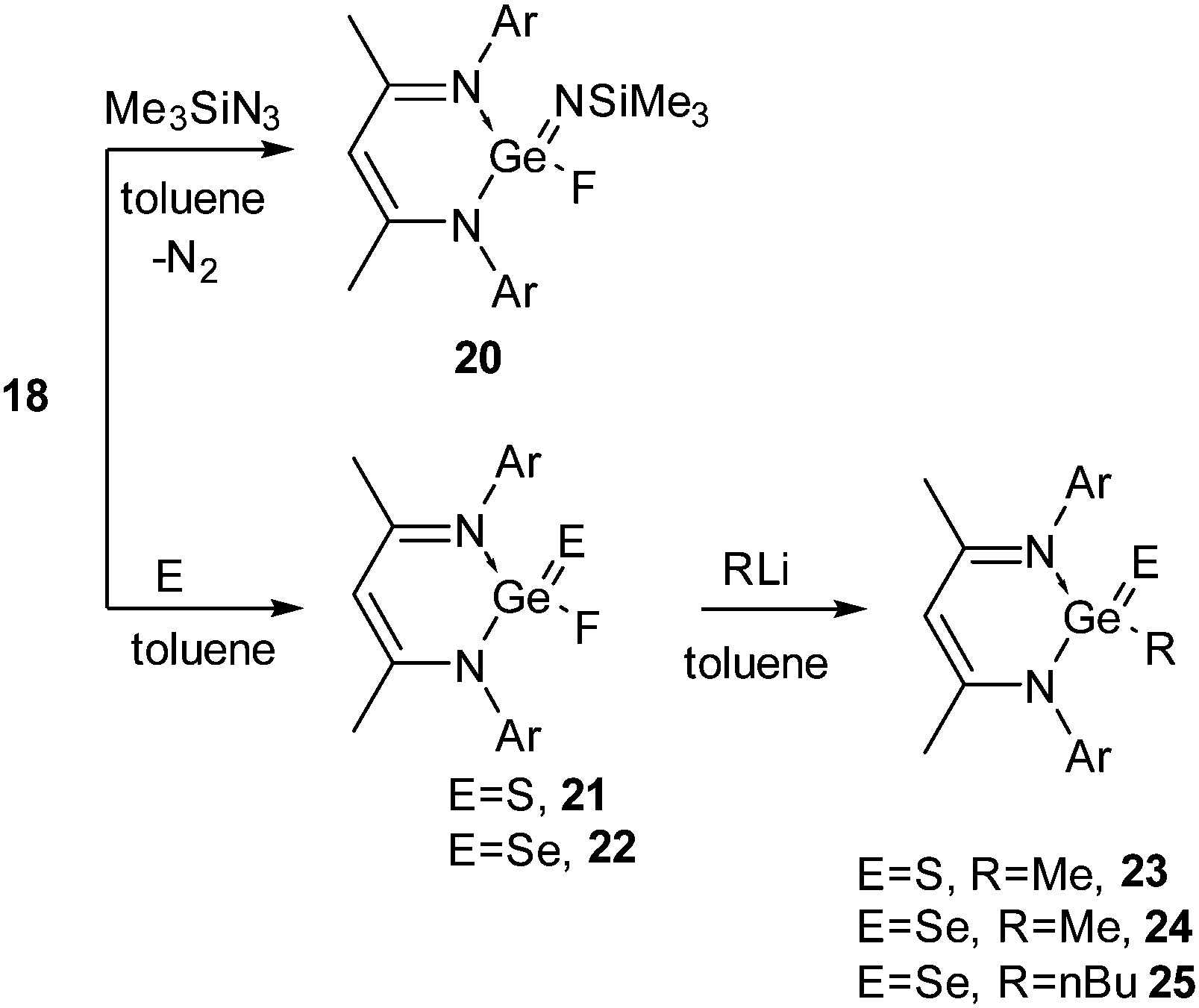 | ||
| Scheme 7 The oxidation reactions of Ge(II) fluoride 18 with sulfur and azide (Ar = 2,6-iPr2C6H3). | ||
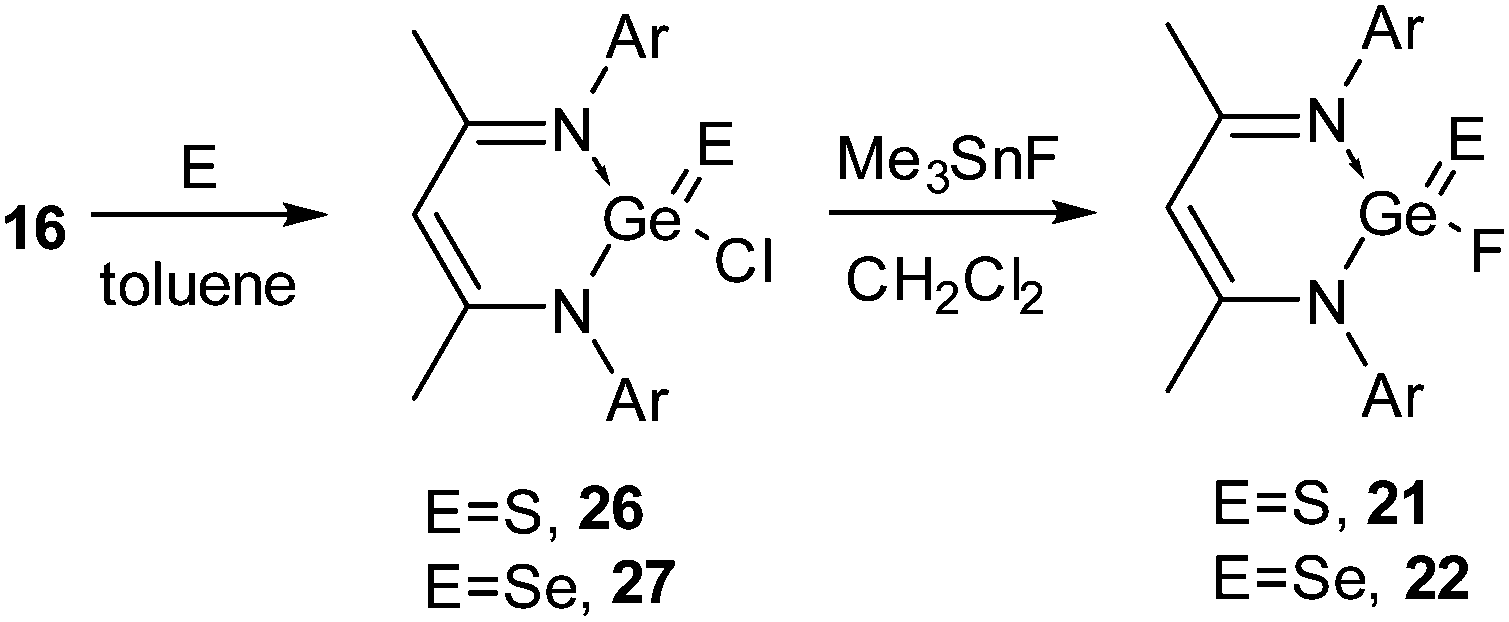 | ||
| Scheme 8 The alternative synthesis of 21 and 22 (Ar = 2,6-iPr2C6H3). | ||
The metathesis between the group 14 chlorides and trimethyltin fluoride was not successful towards synthesizing the corresponding Sn(II) fluorides and no reaction was observed between L1SnCl (28) and Me3SnF even after refluxing for several days. Therefore, the synthetic protocol was modified. Treatment of 28 with MeLi afforded L1SnMe (29), which was subsequently converted to the desired compound, 30 upon refluxing with Me3SnF in toluene for 3 h (Scheme 9).36 The 19F NMR spectrum of 30 shows a singlet at δ −125.29 ppm and the 119Sn NMR spectrum exhibits a doublet at δ −371.52 ppm. The existence of 30 as a monomer was corroborated by single crystal X-ray diffraction studies. The terminal Sn–F bond in 30 was determined to be 1.988(2) Å, which was shorter when compared to the bridging Sn–F–Sn (2.156 (3) Å) bond in trans-[Sn(μ-F)2NC(Me2)(CH2)3CMe2]2 reported by Lappert's group.37
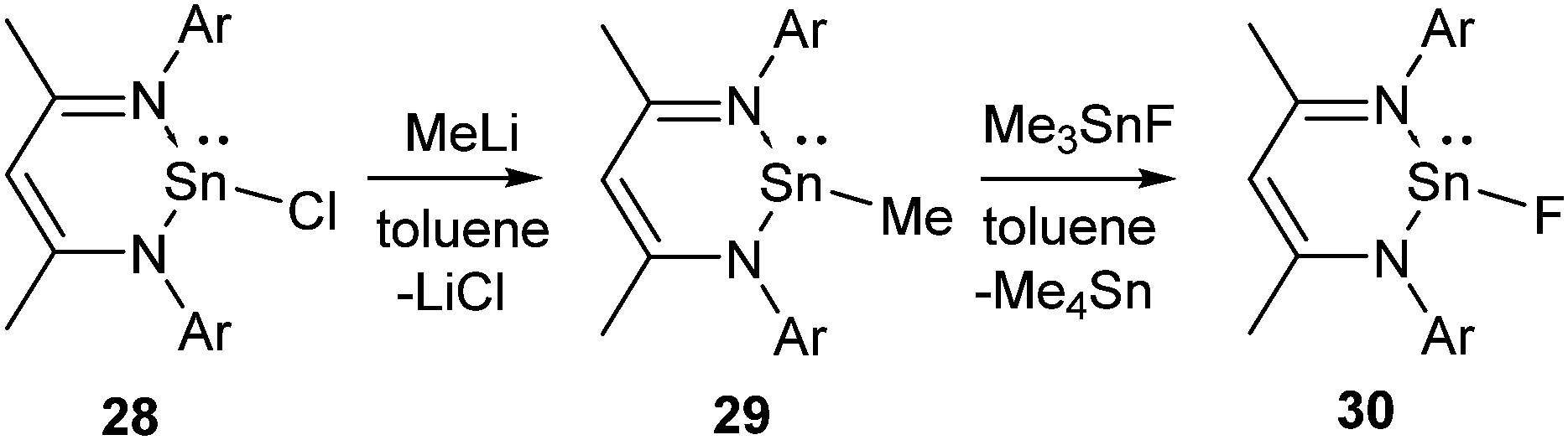 | ||
| Scheme 9 The synthesis of Sn(II) fluoride (Ar = 2,6-iPr2C6H3). | ||
The complexation reactions of Ge(II) and Sn(II) compounds with terminal fluoride atoms are not known. In order to shed light in this area, 18 and 30 were reacted with diiron nonacarbonyl, Fe2(CO)9, at room temperature resulting in the iron carbonyl complexes of germanium(II), L1Ge(F)Fe(CO)4 (31) and tin(II), L1Sn(F)Fe(CO)4 (32), respectively (Scheme 10).38 The Ge–Fe and Sn–Fe bonds were dative in nature. The Ge–Fe bond length in 31 (2.3262(7) Å) is comparable to that in L1Ge(OH)Fe(CO)439 (2.330(1) Å). Similarly, the Sn–Fe bond distance in 32 was found to be 2.4577(6) Å, which is comparable to that in L1Sn(OH)Fe(CO)4 (2.4832(7) Å).40 The Ge–F bond length in 31 (1.868(2) Å) was found to be longer than that in 18 (1.805(17) Å) and 21 (1.848(2) Å), while the Sn–F bond length was reduced in 32 (1.9497(19) Å) than that present in 30 (1.988(2) Å).
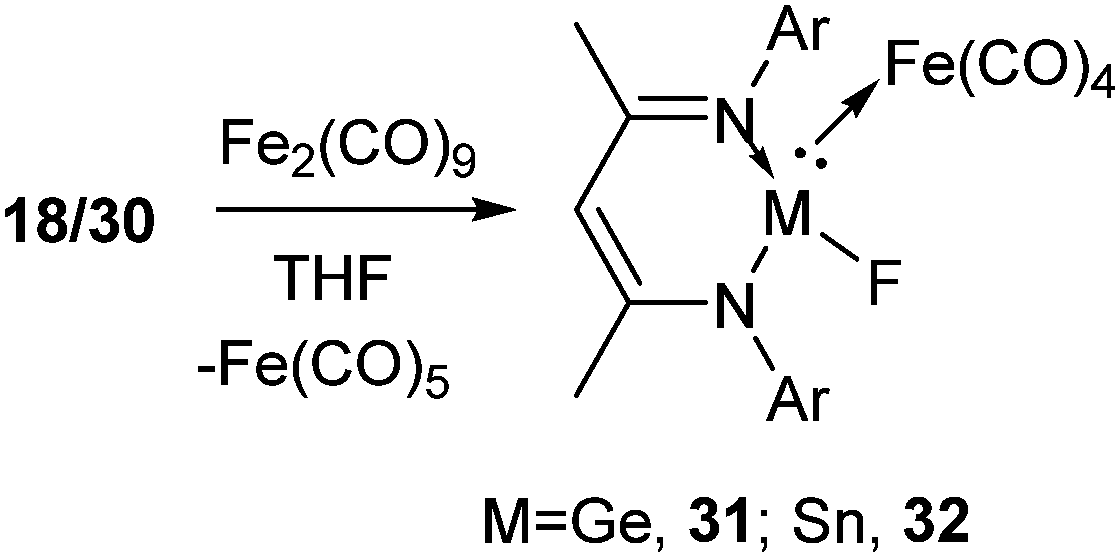 | ||
| Scheme 10 The reactions of 18 and 30 with Fe2(CO)9 (Ar = 2,6-iPr2C6H3). | ||
We have mentioned the use of Pb(II) fluoride before (vide supra, Scheme 2) as a fluorinating agent for the metathesis reaction of Si(II) chloride to fluoride. However, the preparation of Pb(II) fluoride was found to be more challenging than its lighter congeners, which could be attributed to the insolubility associated with the lead compounds. The synthetic strategies applied for the preparation of Ge(II) and Sn(II) fluorides did not furnish the analogous Pb(II) fluoride, and instead, the formation of metallic lead was observed in each reaction. These findings left an impression that Me3SnF should be substituted by another mild fluorinating agent. Moreover, the by-product formed while using Me3SnF as a fluorinating agent is Me3SnCl, which is highly toxic. Therefore, pentafluoropyridine was chosen as a fluorinating agent and reacted with L1Pb(II)NMe2 (33) to afford the β-diketiminatolead(II) monofluoride, L1Pb(II)F (4), together with 4-dimethylaminotetrafluoropyridine (Scheme 11).20 The driving force of this reaction is the formation of a C–N bond, which compensated the energy required for the C–F bond cleavage. The Pb–F bond length in 4 (2.088(17) Å) is expectedly longer than those of Ge–F (1.805(17) Å) in 18 and Sn–F (1.988(2) Å) in 30. The low temperature 19F NMR spectrum of 20 exhibits a resonance at δ 102.7 ppm, accompanied by two 207Pb satellites (δ 106.4 and 99.0 ppm). The low temperature 207Pb NMR spectrum shows a doublet at δ 787.4 ppm with a coupling constant of 1J(19F–207Pb) = 2792 Hz. When the measurements were performed at room temperature, the satellite signals and coupling constants of the 19F and 207Pb NMR spectra were not observed, reflecting the kinetically labile nature of the Pb–F bond at room temperature. The reactive nature of the Pb–F bond was further confirmed when 4 was treated with trifluoroacetophenone to yield a homoleptic Pb(II) compound (34) along with the formation of insoluble PbF2 (Scheme 12).20 The solid-state structure of 34 shows a spirocyclic structure where the two six-membered C3ONPb rings are inter-connected by the common Pb atom.
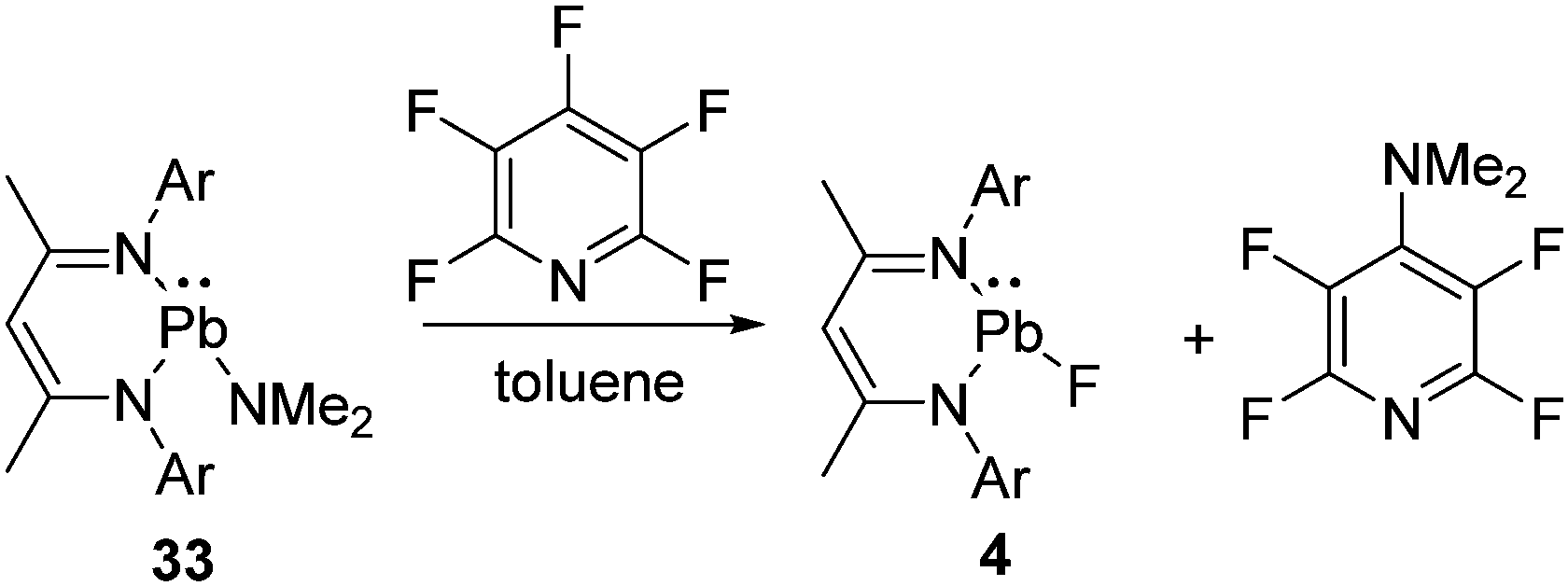 | ||
| Scheme 11 The synthesis of Pb(II) fluoride (Ar = 2,6-iPr2C6H3). | ||
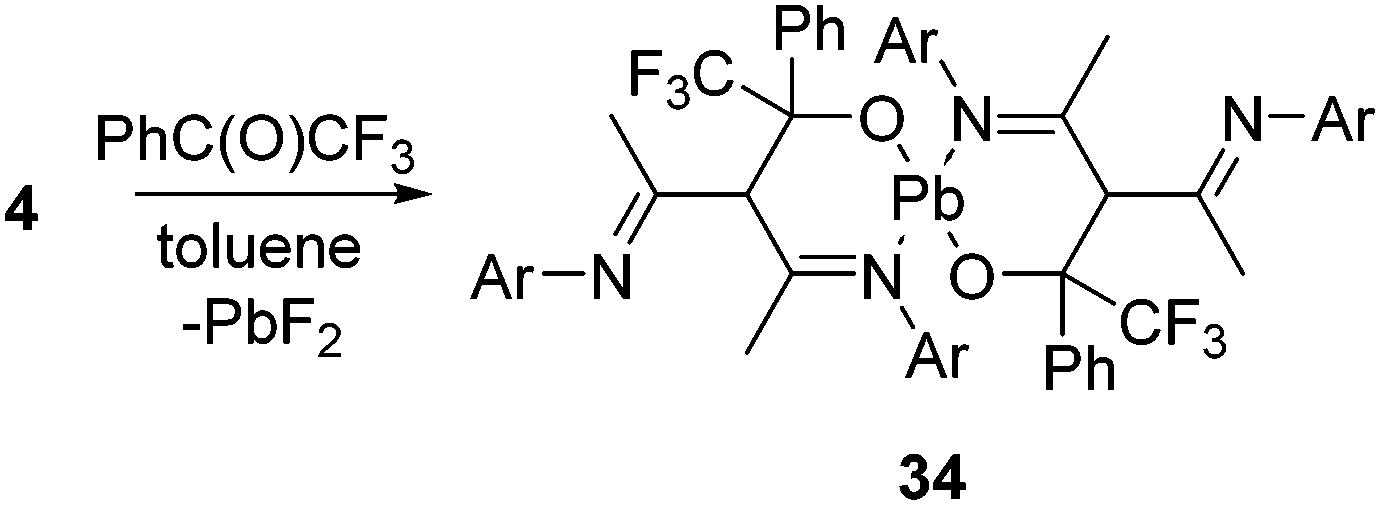 | ||
| Scheme 12 The reaction of 4 with trifluoroacetophenone (Ar = 2,6-iPr2C6H3). | ||
Activation of C–F bonds using compounds with a low valent silicon atom
The activation of C–F bonds in fluorinated hydrocarbons is of fundamental interest from the standpoint of the potential application of organofluorine compounds in synthetic organic chemistry, pharmacy and agrochemistry,41 as well as the ever-increasing environmental concerns related to fluorinated compounds.42 Fluorinated hydrocarbons are not only contributing to global warming but cause the depletion of the ozone layer.42 As a result, there is a need to develop new synthetic strategies for the activation of C–F bonds. Owing to the strength and the resulting high activation barrier of the C–F bonds in fluorocarbons, a strong Lewis acid with high fluoride affinity is sought-after for C–F bond activation. Recent years have witnessed the C–F bond activation using compounds with low-valent main group elements.43–45 For example, the groups of Kuhn, Bertrand, Turner, Lee, Baker, Chaplin, Radius, and many others have demonstrated the activation of aromatic C–F bonds using various carbenes.43 Studer and co-workers have described C–F bond activation in perfluoroarenes using aryl and alkyl isonitriles under UV irradiation.44 The groups of Mikami, Nikonov, and Stephan have independently reported the respective use of boryl lithium, Al(I) compound and phosphonium cations for C–F bond activation.45Compounds with low valent silicon atoms are especially appealing for C−F bond activation because they can integrate both Lewis acidity and fluoride affinity. The propensity of compounds with low valent silicon atom towards oxidative addition reactions can be tapped for cleaving inert bonds such as C–F bonds. Although never accredited, it was the groups of Timms and Margrave who noted that the reaction of hexafluorobenzene and in situ generated SiF2 gave pentafluorophenyltrifluorosilane (C6F5SiF3) as the major product arising from the oxidative addition of one of the C–F bonds at the Si(II) center (Scheme 1, eqn (g)). They were also able to trace the formation of C6F4(SiF3)2 as minor products in an o![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :m:p ratio of ∼1:9:6. This was perhaps the first example of C–F bond activation using a compound with a low valent silicon atom.
:m:p ratio of ∼1:9:6. This was perhaps the first example of C–F bond activation using a compound with a low valent silicon atom.
We embarked our investigation upon the reaction of 1 with C6F6, which led to the oxidative addition of the Si(II) center with the simultaneous cleavage of one of the C–F bonds in C6F6 (Scheme 13).46 Similar activation of the aromatic C–F bond was achieved with C6F5CF3 and C6F5N.46 The resonances of the Si–F moiety present in 35, 36, and 37 arise at δ −63.4, −63.7, and −64.4 ppm in their respective 19F NMR spectra. The 29Si NMR spectra of 35, 36, and 37 show doublets (δ −91.9, −97.2, and −97.9 ppm) with a coupling constant of 1J(29Si–19F) 282.9, 283.6, and 282.7 Hz, respectively.
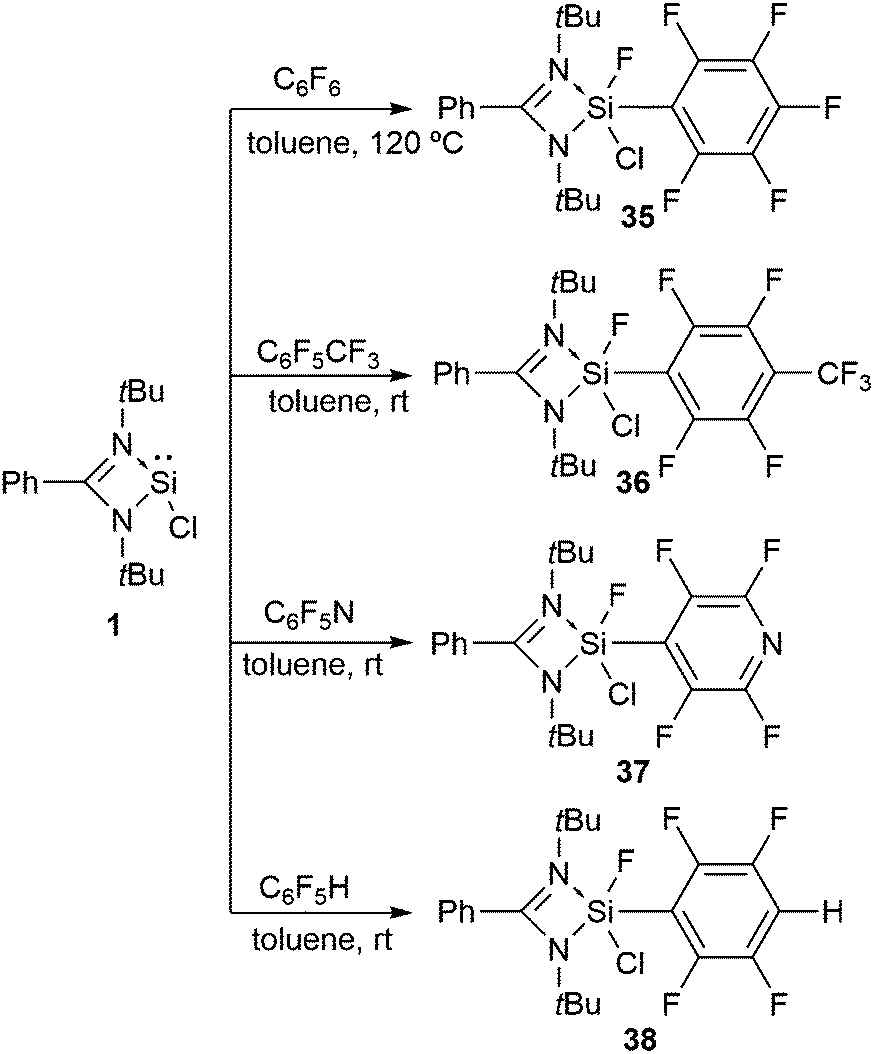 | ||
| Scheme 13 The activation of an aromatic C–F bond using chlorosilylene 1. | ||
The introduction of C–H bonds into the fluorinated substrate often slows down the reactions and generates competition between the C–F and C–H oxidative addition reactions. For example, the Johnson group described the reaction of Ni(PEt3)2 with C6F5H, where the C–H activated species was formed as the kinetic product, while C–F oxidative addition led to the thermodynamic product.47 The reaction of 1 with partially fluorinated benzene, C6F5H, yielded exclusively the product of C–F oxidative addition, 38 (Scheme 13). The formation of the Si–F bond in the product is assumed to drive the C–F bond activation.46
The reactions of the analogous silylene, LSiN(SiMe3)2 (L = PhC(NtBu)2) (39), with hexafluorobenzene and octafluorotoluene follow an analogous oxidative addition pathway (Scheme 14) leading to 40 and 41.48 In the 19F NMR spectra of 40 and 41, the Si–F resonances appear at δ −89.09 and −85.09 ppm, respectively. The 29Si NMR spectrum displays doublets at δ −62.17 ppm for 40 and −71.28 ppm for 41 with a coupling constant of 1J(29Si–19F) 243.19 and 245.02 Hz, respectively.
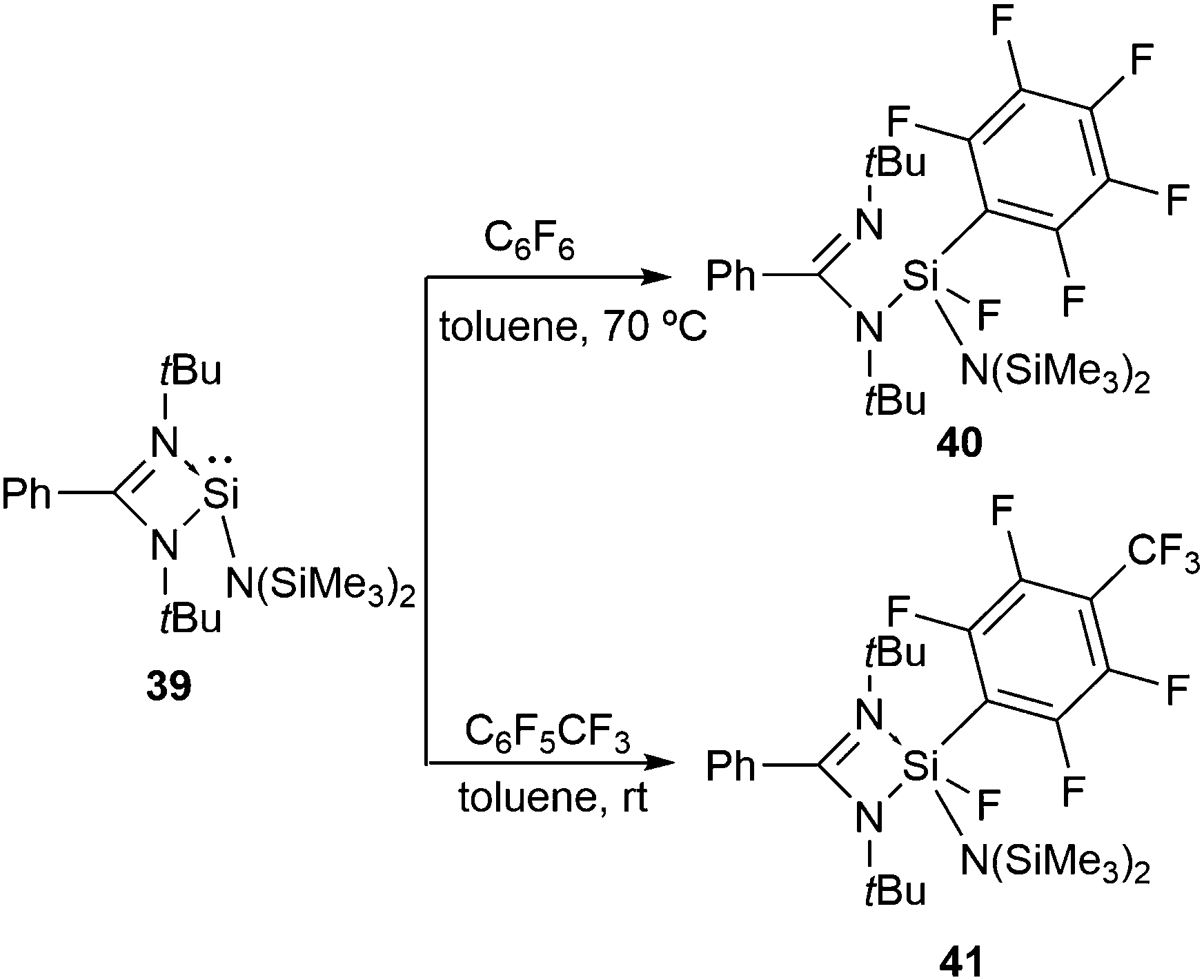 | ||
| Scheme 14 PhC(NtBu)2SiN(SiMe3)2 mediated aromatic C–F bond activation. | ||
Of particular importance is the preference for the activation of the C–F bond para to the CF3 moiety in octafluorotoluene. In a subsequent paper, Koley and co-workers computationally demonstrated that the activation energy barrier for the para C–F bond is lower than the ortho and meta C–F bonds by 5.2 kcal mol−1 and 8.0 kcal mol−1, respectively.49 Moreover, the overall distortion energy for the para C–F bond activation (25.4 kcal mol−1) was found to be significantly smaller than those for the ortho (56.6 kcal mol−1) and meta (63.8 kcal mol−1) C–F bonds. The fact that the activation of the C–F bond in hexafluorobenzene occurs at elevated temperature while in octafluorotoluene its takes place at room temperature can be ascribed to the diminishing strength of the C–F bond upon increasing the fluorination and thereby lowering the energy barriers for the C–F bond activation in the more fluorinated substrates.
Apart from functionalized three coordinate silylenes, such as 1 and 39, typical N-heterocyclic silylene, CH[{(CCH2)(CMe)(2,6-iPr2C6H3N)2}]Si (42), was also reported for the activation of the aromatic C–F bonds.46 All these aforementioned reactions with fluorinated substrates were investigated with 42 (Scheme 15). The oxidative addition of the C–F bond at the Si(II) atom takes place in the reactions with hexafluorobenzene, octafluorotoluene and pentafluoropyridine leading to the formation of 43–45, respectively. However, the reaction of 42 with C6F5H exclusively led to the C–H activated product 46 instead of the C–F activated product observed in the case of 38. No activation of the C–F bond was observed even at low temperature. These differences are likely to be due to the lower coordination number of the Si(II) atom in 42 than that in 1. Usually, silicon hydrides with a high coordinate silicon atom are less stable than those of the corresponding fluoride analogues, which could be the underlying rationality for the preferred formation of low coordinate silicon with hydrides and not with fluorides.
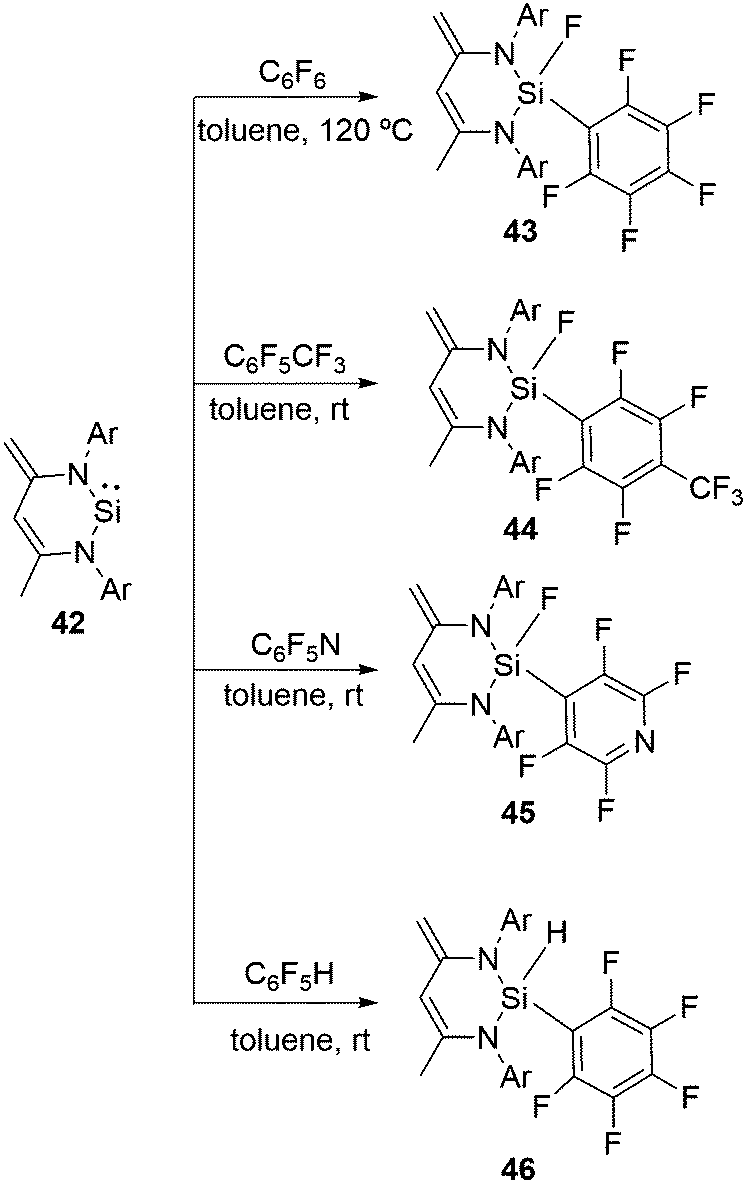 | ||
| Scheme 15 N-Heterocyclic silylene mediated aromatic C–F bond activation (Ar = 2,6-iPr2C6H3). | ||
The selective activation of the aliphatic C(sp3)−F bonds, such as the functionalization of the trifluoromethyl group, was not known for a long time.50 Recently, we have shown that silylene 39 selectively activates one of the C–F bonds in 1,1,1 trifluoro acetophenone leading to a difluorinated alkene (47) featuring both the Si–O and Si–F bond (Scheme 16).48 The DFT calculations revealed that the reaction goes through an Si–O bond formation with an energy barrier (ΔG) of 16.2 kcal mol−1. Subsequently, the reaction proceeded through a five-membered transition state with a corresponding energy barrier of 6.8 kcal mol−1, where a Si–F bond and CC bond are formed. The final product, 47, is very thermodynamically stable (−41.7 kcal mol−1), which, along with the low barriers obtained, indicates that this reaction is both kinetically and thermodynamically viable at room temperature. Insertion of a Si(II) atom into the CF3 group of hexafluoroacetimine was also reported. The reactions of 1 and 42 with PhNC(CF3)2 led to the facile C–F activation of one of the CF3 substituents and the formation of difluorinated alkenes, such as 48 and 49 (Scheme 17).51 Such selective activation of one of the C–F bonds rather than complete defluorination of the CF3 group by compounds with low valent silicon atoms was not known beforehand.
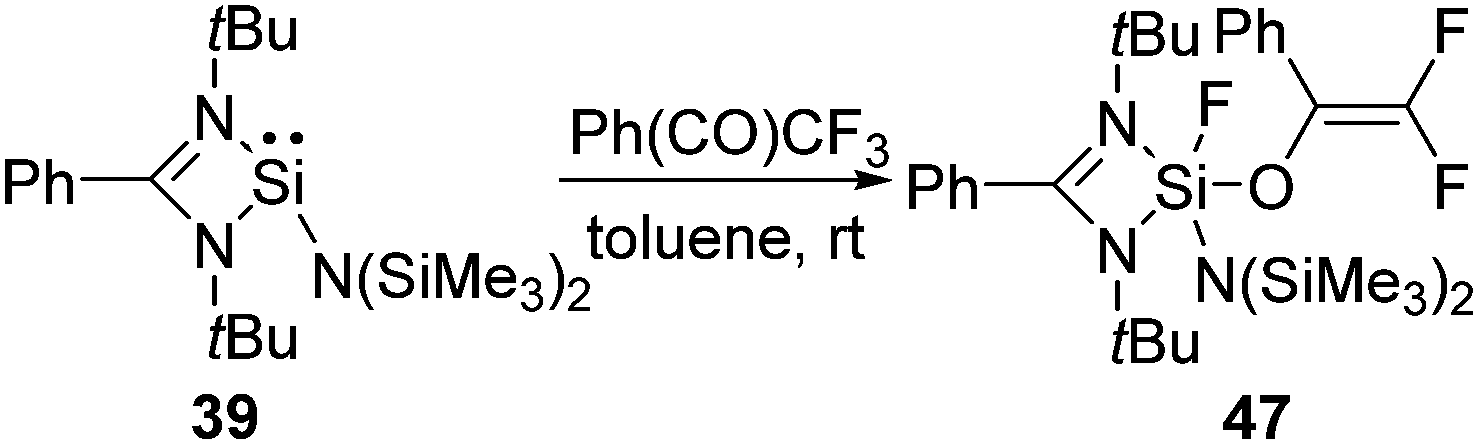 | ||
| Scheme 16 Activation of the C–F bond in trifluoro acetophenone by 39. | ||
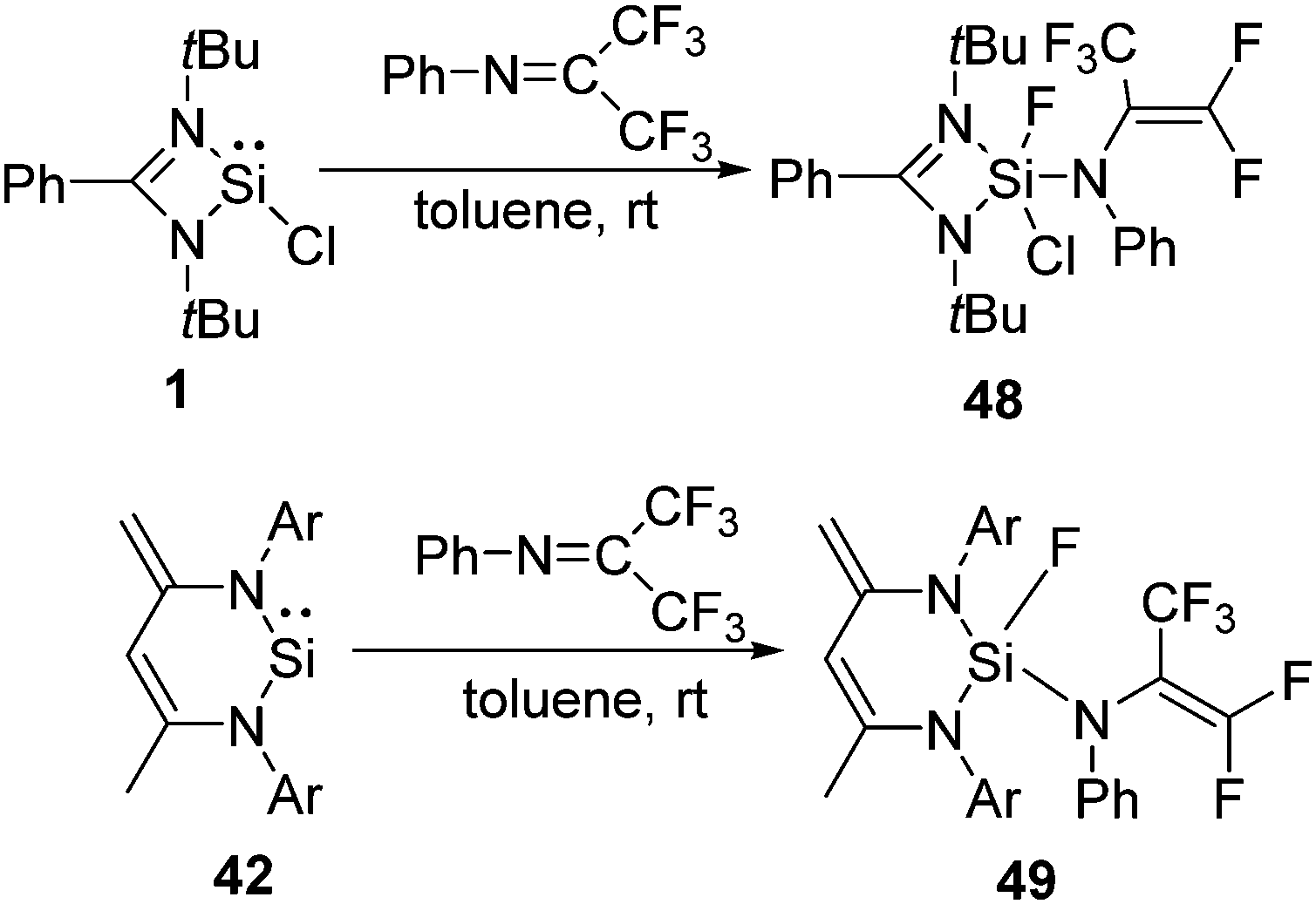 | ||
| Scheme 17 Activation of the C–F bond in PhCN(CF3)2 by 1 and 42. | ||
Activation of C–F bonds using compounds with low valent germanium and tin atoms
In addition to silylenes, germylenes and stannylenes have also been found to activate aromatic C–F bonds. In an early example, we showed that both {PhC(NtBu)2}GeNiPr2(50) and {PhC(NtBu)2}SnNMe2 (51) can activate the C–F bond in pentafluoropyridine, however, divergent behavior was observed regarding their reactivities.52 Like in the cases of silylenes, the C–F bond of pentafluoropyridine underwent an oxidative addition reaction at the Ge(II) atom leading to the formation of 52. However, no oxidative addition product was formed with 51 and instead the substitution of the NMe2 group by the fluoride ion occurred at the Sn(II) atom resulting in 53 (Scheme 18). Single crystal X-ray diffraction studies on 53 revealed that it exists as a dimer in which the two molecules are connected by weak intermolecular Sn⋯F bonds.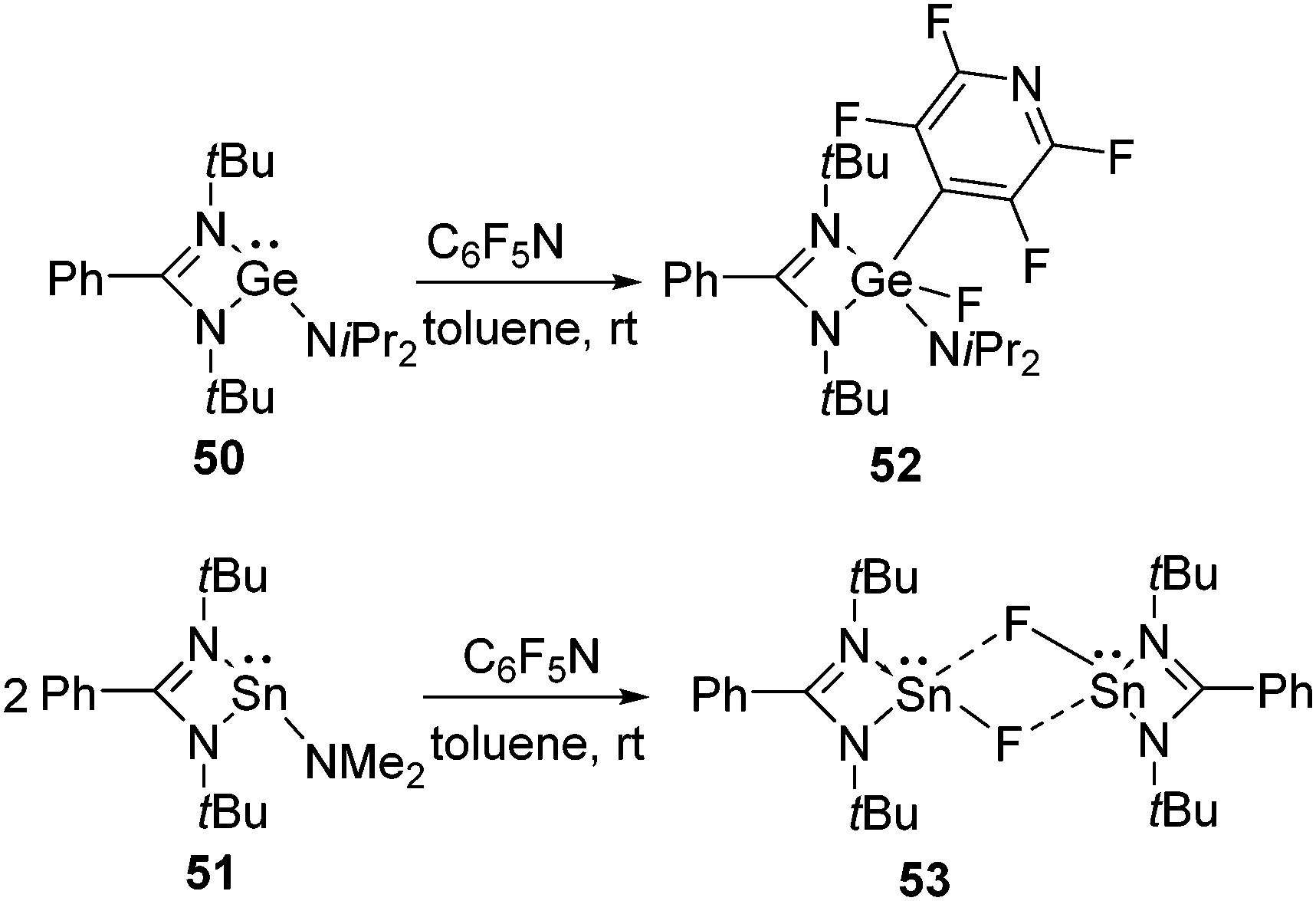 | ||
| Scheme 18 Activation of C–F bond of C6F5N by germylene and stannylene. | ||
In a subsequent theoretical paper, Koley and co-workers explained the dichotomous reactivity pattern of germylene 50 and stannylene 51 towards pentafluoropyridine.49 In 50, the electron density is located on the Ge atom and due to the presence of the NiPr2 substituent on Ge, the transition state for the oxidative addition pathway was found to be more favored than the metathesis pathway by 32.1 kcal mol−1. However, they predicted that substitution on the Ge atom plays a part in determining which product will be formed. For example, they calculated that the replacement of the NiPr2 substituent with an NMe2 group may lead to a competition between the oxidative addition and metathesis pathways as the electronic and steric environments support both the transition states. An experimental verification of this prediction is deemed desirable and may open up a new avenue for C–F bond activation using functionalized germylenes. However, in case of 51, the HOMO–LUMO energy gap increases due to the larger s–p gap. Therefore, the metathesis reaction was found to be more favored over the oxidative addition reaction by 13.3 kcal mol−1. The same group also demonstrated the lower HOMO–LUMO energy gap is required for para C–F bond activation than the corresponding ortho or meta C–F bond activations and thereby explained the penchant for the para C–F bond activation of pentafluoropyridine.
Tin(II) hydride 54 was also found to activate the C–F bond of C6F6 under ambient conditions and resulted in tin(II) fluoride 30 along with the tin(II) pentafluorophenyl compound, 55, and C6F5H (Scheme 19).53 A closer look at this reaction revealed that stoichiometric hydrodefluorination of C6F6 to C6F5H takes place during the reaction. The reaction is likely to occur via the metathesis between hydride and fluoride as well as hydride and the pentafluorophenyl substituent. The formation of 55 during the reaction was indicated by the 119Sn NMR resonance at δ −176.4 ppm, which is significantly high-field shifted with respect to that of 54 (δ −4.5 ppm, 1JSn–H = 64 Hz)54 and 30 (δ −371.5 ppm, 1JSn–F = 3100 Hz). To unequivocally prove the formation of 55 during the reaction, 28 was reacted with C6F5Li, which exclusively led to 55 and the resulting NMR spectra were compared.
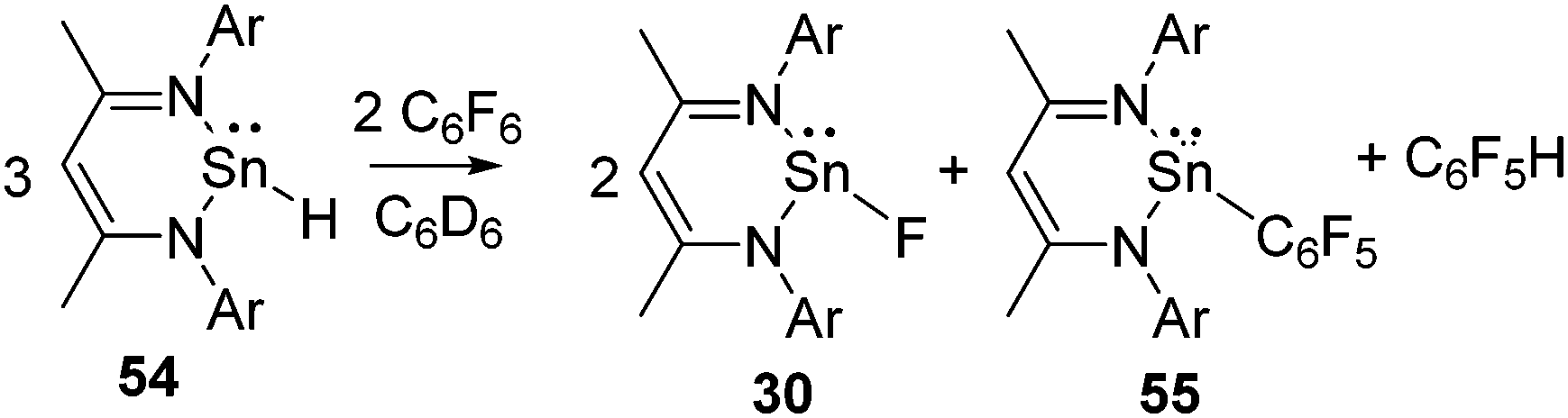 | ||
| Scheme 19 Activation of C–F bond of C6F6 by stannylene hydride. | ||
Silylium ion in the hydrodefluorination reaction
Hydrodefluorination is a reaction in which a C–F bond is replaced by a C–H bond. The removal of fluorines from fluorocarbons compounds and replacement with hydrogen is an important goal, in order to convert them into more biodegradable compounds. Usually, transition metal catalysts are used for this conversion.55 Ozerov and co-workers showed that a silylium–borane catalyst, Et3Si+[B(C6F5)4]−, can catalyze the selective hydrodefluorination of C6F5CF3 to C6F5CH3, although with a poor turn over number (TON = 19).56 By varying the counter-anion with a carborane catalyst, Et3Si+[CHB11H5Cl6]−, the TON of the same reaction was found to increase up to 2700 (Scheme 20).57 The mechanism has been assumed to proceed via a silylium ion-mediated fluoride abstraction from the substrate with subsequent hydride transfer from the organosilane to the intermediate carbenium ion. The driving force for the overall reaction is the formation of Si–F and C–H bonds that counterbalance the cleavage of the C–F and Si–H bonds by 45.4 kcal mol−1, thereby enabling the metathesis.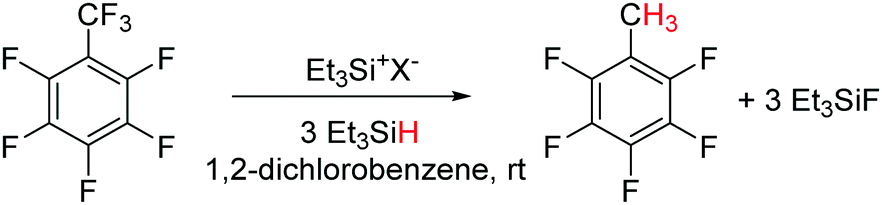 | ||
| Scheme 20 Silylium ion catalyzed hydrodefluorination. | ||
Parallel to Ozerov's findings, Müller and co-workers reported the hydrodefluorination of trifluorotoluene to give toluene using a hydride-bridged disilyl cation as the catalyst (Scheme 21).58 The specialty of this case is that the cation combines the silicon electrophile as well as the hydride source. Both hydride (I) and fluoride (II) bridged silylium cations were observed as intermediates, structurally characterized and found to be interconvertible in the reaction with alkylfluorides or silanes. The tentative catalytic cycle is shown in Scheme 21, however, the cycle operates with a moderate TON.
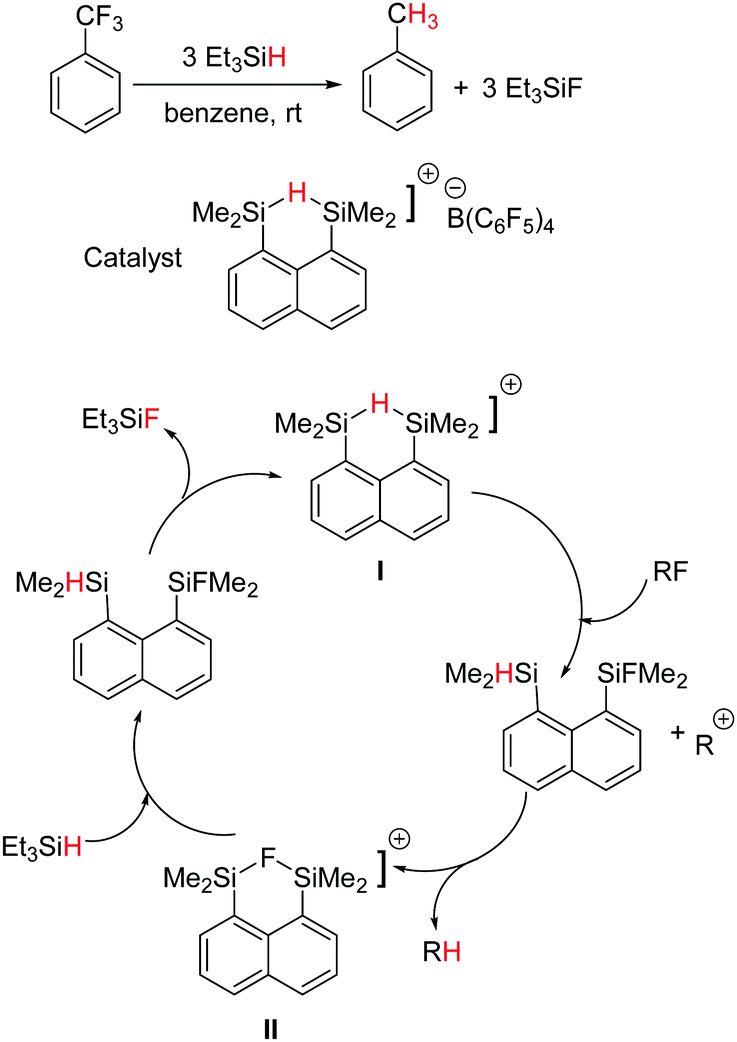 | ||
| Scheme 21 Silylium ion catalysed hydrodefluorination of trifluorotoluene and the tentative catalytic cycle. | ||
Conclusions
While the basis of silicon(II) difluoride chemistry dates back into the 1970s, most progress in this area has been made during the last few years. The first stable silicon(II) difluoride (14) was made accessible using cyclic alkyl amino carbene (cAAC) ligand. The synthesis of the first compound containing an SiF3 radical (15) has also been recently accomplished. The lion's share of the credit must go to the cAAC ligand due to its high lying HOMO and low lying LUMO, which not only donates electron density to the Si(II) center but unlike NHC, can accept electron density from the Si–F bonds. Structural analysis of these complexes in tandem with DFT calculations has significantly enhanced the understanding of their electronic properties. The reactivities of both 14 and 15 have yet to be reported but promise rich further chemistry. And of course, the success of cAAC and the failure of NHC (so far!!) for stabilizing the SiF2 moiety will encourage synthetic chemists to make newer neutral ligands with tunable electronic properties. The chemistry of silicon(II) fluorides can be best described in Churchill's words “…it is not even the beginning of the end…”. The game is on!Of equal importance are the recent achievements in the activation of C–F bonds using compounds with low valent silicon atoms. Progress in the use of silylenes to activate both alkyl and aryl C–F bonds was also detailed. The formation of a strong Si–F bond is likely to be the driving impetus of these reactions. In addition to the commonly observed stoichiometric C–F bond activation, silylium ion catalyzed hydrodefluorination has been discovered with remarkable turn over numbers, which was previously considered the exclusive domain of transition metal complexes. Apart from the hydrodefluorination reactions, silylium cations have been recently employed in the Friedel Crafts reactions of fluoroarenes59 and C–C coupling reactions,60 which have not been discussed in this feature article. As a consequence, the focus in silicon fluorine chemistry will likely tilt from fundamental to application-oriented research in the near future. As part of a natural progression, the heavier congeners of silylenes, such as germylenes and stannylenes, have also been reported for C–F bond activation but their examples are still very scant. Nevertheless, we hold great hopes for studying these riveting molecules in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
SSS thanks SERB (SB/S2/RJN-073/2014), India and HWR thanks DFG (RO 224/68-1), Germany for financial support. We warmly thank our students and postdoctoral co-workers, whose names are cited in the references, for their many intellectual and experimental contributions.Notes and references
- R. West, M. J. Fink and J. Michl, Science, 1981, 214, 1343–1344 CAS.
- (a) I. Alvarado-Beltran, A. Rosas-Sanchez, A. Baceiredo, N. Saffon-Merceron, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2017, 56, 10481–10485 CrossRef CAS PubMed; (b) D. Wendel, D. Reiter, A. Porzelt, P. J. Altmann, S. Inoue and B. Rieger, J. Am. Chem. Soc., 2017, 139, 17193–17198 CrossRef CAS PubMed.
- For selected references on important discoveries on compounds with low valent silicon, please see: (a) M. Kira, S. Ishida, T. Iwamoto and C. Kabuto, J. Am. Chem. Soc., 1999, 121, 9722–9723 CrossRef CAS; (b) S. S. Sen, A. Jana, H. W. Roesky and C. Schulzke, Angew. Chem., Int. Ed., 2009, 48, 8536–8538 CrossRef CAS PubMed; (c) A. C. Filippou, O. Chernov and G. Schnakenburg, Angew. Chem., Int. Ed., 2009, 48, 5687–5690 CrossRef CAS PubMed; (d) Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer III, P. V. R. Schleyer and G. H. Robinson, Science, 2008, 321, 1069–1071 CrossRef CAS PubMed; (e) A. Sekiguchi, R. Kinjo and M. Ichinohe, Science, 2004, 305, 1755–1757 CrossRef CAS PubMed; (f) K. Abersfelder, A. J. P. White, H. S. Rzepa and D. Scheschkewitz, Science, 2010, 327, 564–566 CrossRef CAS PubMed; (g) S. S. Sen, H. W. Roesky, K. Meindl, D. Stern, J. Henn, A. C. Stückl and D. Stalke, Chem. Commun., 2010, 46, 5873–5875 RSC; (h) S. S. Sen, J. Hey, M. Eckhardt, R. Herbst-Irmer, E. Maedl, R. A. Mata, H. W. Roesky, M. Scheer and D. Stalke, Angew. Chem., Int. Ed., 2011, 50, 12510–12513 CrossRef CAS PubMed; (i) A. V. Protchenko, K. H. Birjkumar, D. Dange, A. D. Schwarz, D. Vidovic, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 6500–6503 CrossRef CAS PubMed.
- W. Kutzelnigg, Angew. Chem., Int. Ed. Engl., 1984, 23, 272–295 CrossRef.
- For reviews in compounds with low valent silicon atoms and related compounds, please see: (a) M. Haaf, T. A. Schmedake and R. West, Acc. Chem. Res., 2000, 33, 704–714 CrossRef CAS PubMed; (b) S. Nagendran and H. W. Roesky, Organometallics, 2008, 27, 457–492 CrossRef CAS; (c) Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2009, 109, 3479–3511 CrossRef CAS PubMed; (d) M. Kira, Chem. Commun., 2010, 46, 2893–2903 RSC; (e) S. K. Mandal and H. W. Roesky, Chem. Commun., 2010, 46, 6016–6041 RSC; (f) M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed; (g) S. S. Sen., S. Khan, S. Nagendran and H. W. Roesky, Acc. Chem. Res., 2012, 45, 578–587 CrossRef CAS PubMed; (h) S. S. Sen., S. Khan, P. P. Samuel and H. W. Roesky, Chem. Sci., 2012, 3, 659–682 RSC; (i) S. Yadav, S. Saha and S. S. Sen, ChemCatChem, 2016, 8, 486–501 CrossRef CAS.
- (a) K. Reimer and F. Tiemann, Eur. J. Inorg. Chem., 1876, 1268–1278 Search PubMed; (b) J. U. Nef, Justus Liebigs Ann. Chem., 1895, 287, 265 CrossRef CAS; (c) J. U. Nef, Justus Liebigs Ann. Chem., 1892, 289, 202 Search PubMed.
- (a) E. G. Rochow, Silicon and Silicones, Springer Verlag, 1987 Search PubMed; (b) J. Teichmann and M. Wagner, Chem. Commun., 2018, 54, 1397–1412 RSC.
- (a) M. Schmeißer and M. Schwarzmann, Z. Naturforsch., B: J. Chem. Sci., 1956, 11, 278 Search PubMed; (b) H. Bloching, PhD thesis, Freie Universität, Berlin, 1961.
- (a) D. A. J. Osteraas, J. Appl. Polym. Sci., 1969, 13, 1523–1535 CrossRef; (b) K. Hirai, T. Itoh and H. Tomioka, Chem. Rev., 2009, 109, 3275–3332 CrossRef CAS PubMed; (c) N. Auner, Synthetic Methods of Organometallic and Inorganic Chemistry, Thieme, 1996, vol. 2 Search PubMed.
- M. Schmeißer, Angew. Chem., 1954, 66, 713 CrossRef.
- (a) P. L. Timms, T. C. Ehlert and J. L. Margrave, J. Am. Chem. Soc., 1965, 87, 3819–3823 CrossRef CAS; (b) P. L. Timms, D. D. Stump, R. A. Kent and J. L. Margrave, J. Am. Chem. Soc., 1966, 88, 940–942 CrossRef CAS; (c) J. C. Thompson and J. L. Margrave, Science, 1967, 155, 669–671 CrossRef CAS PubMed.
- S. Sinhababu, S. Kundu, A. N. Paesch, R. Herbst-Irmer, D. Stalke, I. Fernandez, G. Frenking, A. C. Stückl, B. Schwederski, W. Kaim and H. W. Roesky, Chem. – Eur. J., 2018, 24, 1264–1268 CrossRef CAS PubMed.
- D. C. Pease, US Pat., 2840588 and 3026173, 1958, assigned to the Du Pont Co., Wilmington, Del Search PubMed.
- M. Schmeißer and K.-P. Ehlers, Angew. Chem., Int. Ed., 1964, 3, 700 CrossRef.
- P. L. Timms, R. A. Kent, T. C. Ehlert and J. L. Margrave, J. Am. Chem. Soc., 1965, 87, 2824–2828 CrossRef CAS.
- (a) T. C. Ehlert and J. L. Margrave, J. Chem. Phys., 1964, 41, 1066–1072 CrossRef CAS; (b) V. M. Khanna, R. Hauge, R. F. Curl and J. L. Margrave, J. Chem. Phys., 1967, 47, 5031–5036 CrossRef CAS.
- J. L. Margrave and P. W. Wilson, Acc. Chem. Res., 1971, 4, 145–152 CrossRef CAS.
- (a) S. S. Sen, H. W. Roesky, D. Stern, J. Henn and D. Stalke, J. Am. Chem. Soc., 2010, 132, 1123–1126 CrossRef CAS PubMed; (b) R. S. Ghadwal, H. W. Roesky, S. Merkel, J. Henn and D. Stalke, Angew. Chem., Int. Ed., 2009, 48, 5683–5686 CrossRef CAS PubMed.
- P. Jutzi, U. Holtmann, P. Bögge and A. Müller, J. Chem. Soc., Chem. Commun., 1988, 305–306 RSC.
- A. Jana, S. P. Sarish, H. W. Roesky, D. Leusser, I. Objartel and D. Stalke, Chem. Commun., 2011, 47, 5434–5436 RSC.
- H. Braunschweig, R. D. Dewhurst and V. H. Gessner, Chem. Soc. Rev., 2013, 42, 3197–3208 RSC.
- (a) S. M. I. Al-Rafia, A. C. Malcolm, M. J. Ferguson, R. McDonald and E. Rivard, Angew. Chem., Int. Ed., 2011, 50, 8354–8357 CrossRef CAS PubMed; (b) S. M. I. Al-Rafia, A. C. Malcolm, S. K. Liew and E. Rivard, J. Am. Chem. Soc., 2011, 133, 777–779 CrossRef CAS PubMed.
- R. Azhakar, R. S. Ghadwal, H. W. Roesky, H. Wolf and D. Stalke, J. Am. Chem. Soc., 2012, 134, 2423–2428 CrossRef CAS PubMed.
- M. Z. Kassaee, M. R. Momeni, F. A. Shakib, M. Ghambarian and S. M. Musavi, Struct. Chem., 2010, 21, 593–598 CrossRef CAS.
- V. Lavallo, Y. Canac, B. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 CrossRef CAS PubMed.
- (a) K. C. Mondal, S. Roy and H. W. Roesky, Chem. Soc. Rev., 2016, 45, 1080–1111 RSC; (b) S. Roy, K. C. Mondal and H. W. Roesky, Acc. Chem. Res., 2016, 49, 357–369 CrossRef CAS PubMed.
- Y. Ding, H. Hao, H. W. Roesky, M. Noltemeyer and H.-G. Schmidt, Organometallics, 2001, 20, 4806–4811 CrossRef CAS.
- T. Ochiai, T. Szilvási, D. Franz, E. Irran and S. Inoue, Angew. Chem., Int. Ed., 2016, 55, 11619–11624 CrossRef CAS PubMed.
- (a) Y. Ding, Q. Ma, I. Usón, H. W. Roesky, M. Noltemeyer and H.-G. Schmidt, J. Am. Chem. Soc., 2002, 124, 8542–8543 CrossRef CAS PubMed; (b) Y. Ding, Q. Ma, H. W. Roesky, I. Usón, M. Noltemeyer and H.-G. Schmidt, Dalton Trans., 2003, 1094–1098 RSC.
- S. S. Sen, R. S. Ghadwal, D. Kratzert, D. Stern, H. W. Roesky and D. Stalke, Organometallics, 2011, 30, 1030–1033 CrossRef CAS.
- (a) M. Veith, S. Becker and V. Huch, Angew. Chem., Int. Ed. Engl., 1989, 28, 1237–1238 CrossRef; (b) N. Tokitoh, T. Matsumoto, K. Manmaru and R. Okazaki, J. Am. Chem. Soc., 1993, 115, 8855–8856 CrossRef CAS.
- T. Matsumoto, N. Tokitoh and R. Okazaki, Angew. Chem., Int. Ed. Engl., 1994, 33, 2316–2317 CrossRef.
- T. Matsumoto, N. Tokitoh and R. Okazaki, J. Am. Chem. Soc., 1999, 121, 8811–8824 CrossRef CAS.
- S. Sinhababu, R. K. Sitwatch, G. Mukherjee, G. Rajaraman and S. Nagendran, Inorg. Chem., 2012, 51, 9240–9248 CrossRef CAS PubMed.
- H.-S. Kim, E. A. Jung, S. H. Han, J. H. Han, B. K. Park, C. G. Kim and T.-M. Chung, Inorg. Chem., 2017, 56, 4084–4092 CrossRef CAS PubMed.
- A. Jana, H. W. Roesky, C. Schulzke, A. Döring, T. Beck, A. Pal and R. Herbst-Irmer, Inorg. Chem., 2009, 48, 193–197 CrossRef CAS PubMed.
- R. W. Chorley, D. Ellis, P. B. Hitchcock and M. F. Lappert, Bull. Soc. Chim. Fr., 1992, 129, 599–604 Search PubMed.
- A. Jana, P. P. Samuel, H. W. Roesky and C. Schulzke, J. Fluorine Chem., 2010, 131, 1096–1099 CrossRef CAS.
- S. P. Sarish, H. W. Roesky, M. John, A. Ringe and J. Magull, Chem. Commun., 2009, 2390–2392 RSC.
- A. Jana, S. P. Sarish, H. W. Roesky, C. Schulzke and P. P. Samuel, Chem. Commun., 2010, 46, 707–709 RSC.
- (a) J.-P. Begue and D. Bonnet-Delpon, J. Fluorine Chem., 2006, 127, 992–1012 CrossRef CAS; (b) C. Isanbor and D. O’Hagan, J. Fluorine Chem., 2006, 127, 303–319 CrossRef CAS; (c) K. L. Kirk, J. Fluorine Chem., 2006, 127, 1013–1029 CrossRef CAS.
- (a) K. P. Shine and W. T. Sturges, Science, 2007, 315, 1804–1805 CrossRef CAS PubMed; (b) F. S. Rowland, Angew. Chem., Int. Ed. Engl., 1996, 35, 1786–1798 CrossRef CAS.
- (a) N. Kuhn, J. Fahl, R. Boese and G. Henkel, Z. Naturforsch., B: Chem. Sci., 1998, 53, 881–886 CAS; (b) S. Styra, M. Melaimi, C. E. Moore, A. L. Rheingold, T. Augenstein, F. Breher and G. Bertrand, Chem. – Eur. J., 2015, 21, 8441–8446 CrossRef CAS PubMed; (c) Z. R. Turner, Chem. – Eur. J., 2016, 22, 11461–11468 CrossRef CAS PubMed; (d) Y. Kim and E. Lee, Chem. Commun., 2016, 52, 10922–10925 RSC; (e) M. C. Leclerc, S. I. Gorelsky, B. M. Gabidullin, I. Korobkov and R. T. Baker, Chem. – Eur. J., 2016, 22, 8063–8067 CrossRef CAS PubMed; (f) M. C. Leclerc, B. M. Gabidullin, J. G. Da Gama, S. L. Daifuku, T. E. Iannuzzi, M. L. Neidig and R. T. Baker, Organometallics, 2017, 36, 849–857 CrossRef CAS PubMed; (g) J. Emerson-King, S. A. Hauser and A. B. Chaplin, Org. Biomol. Chem., 2017, 15, 787–789 RSC; (h) U. S. D. Paul and U. Radius, Chem. – Eur. J., 2017, 23, 3993–4009 CrossRef CAS PubMed.
- A. Dewanji, C. Mück-Lichtenfeld, K. Bergander, C. G. Daniliuc and A. Studer, Chem. – Eur. J., 2015, 21, 12295–12298 CrossRef CAS PubMed.
- (a) S. Ito, N. Kato and K. Mikami, Chem. Commun., 2017, 53, 5546–5548 RSC; (b) T. Chu, Y. Boyko, I. Korobkov and G. I. Nikonov, Organometallics, 2015, 34, 5363–5365 CrossRef CAS; (c) I. Mallov, T. C. Johnstone, D. C. Burns and D. W. Stephan, Chem. Commun., 2017, 53, 7529–7532 RSC.
- A. Jana, P. P. Samuel, G. Tavčar, H. W. Roesky and C. Schulzke, J. Am. Chem. Soc., 2010, 132, 10164–10170 CrossRef CAS PubMed.
- S. A. Johnson, C. W. Huff, F. Mustafa and M. Saliba, J. Am. Chem. Soc., 2008, 130, 17278–17280 CrossRef CAS PubMed.
- V. S. V. S. N. Swamy, N. Parvin, K. V. Raj, K. Vanka and S. S. Sen, Chem. Commun., 2017, 53, 9850–9853 RSC.
- T. Mondal, S. De and D. Koley, Inorg. Chem., 2017, 56, 10633–10643 CrossRef CAS PubMed.
- H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119–2183 CrossRef CAS PubMed.
- R. Azhakar, H. W. Roesky, H. Wolf and D. Stalke, Chem. Commun., 2013, 49, 1841–1843 RSC.
- P. P. Samuel, A. P. Singh, S. P. Sarish, J. Matussek, I. Objartel, H. W. Roesky and D. Stalke, Inorg. Chem., 2013, 52, 1544–1549 CrossRef CAS PubMed.
- A. Jana, H. W. Roesky, C. Schulzke and P. P. Samuel, Organometallics, 2010, 29, 4837–4841 CrossRef CAS.
- A. Jana, H. W. Roesky, C. Schulzke and A. Döring, Angew. Chem., Int. Ed., 2009, 48, 1106–1109 CrossRef CAS PubMed.
- For selected references on hydrodefluorination by transition metals, please see: (a) J. Vela, J. M. Smith, Y. Yu, N. A. Ketterer, C. J. Flaschenriem, R. J. Lachicotte and P. L. Holland, J. Am. Chem. Soc., 2005, 127, 7857–7870 CrossRef CAS PubMed; (b) T. F. Beltran, M. Feliz, R. Llusar, J. A. Mata and V. S. Safont, Organometallics, 2011, 30, 290–297 CrossRef CAS; (c) D. Breyer, T. Braun and P. Kläring, Organometallics, 2012, 31, 1417–1424 CrossRef CAS; (d) L. M. Guard, A. E. Ledger, S. P. Reade, C. Ellul, M. E. Mahon and M. K. Whittlesey, J. Organomet. Chem., 2011, 696, 780–786 CrossRef CAS; (e) M. F. Kühnel and D. Lentz, Angew. Chem., Int. Ed., 2010, 49, 2933–2936 CrossRef PubMed.
- V. J. Scott, R. Ćelenligil-Ćetin and O. V. Ozerov, J. Am. Chem. Soc., 2005, 127, 2852–2853 CrossRef CAS PubMed.
- O. V. Ozerov and C. Douvris, Science, 2008, 321, 1188–1190 CrossRef PubMed.
- R. Panisch, M. Bolte and T. Müller, J. Am. Chem. Soc., 2006, 128, 9676–9682 CrossRef CAS PubMed.
- O. Allemann, S. Duttwyler, P. Romanato, K. K. Baldridge and J. S. Siegel, Science, 2011, 332, 574–577 CrossRef CAS PubMed.
- B. Shao, A. L. Bagdasarian, S. Popov and H. M. Nelson, Science, 2017, 355, 1403–1407 CrossRef CAS PubMed.
Footnote |
| † Dedicated in the memory of Professor George A. Olah and on the occasion of the 60th birthday of Professor Dr Alexander C. Filippou. |
| This journal is © The Royal Society of Chemistry 2018 |