
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From fundamental supramolecular chemistry to self-assembled nanomaterials and medicines and back again – how Sam inspired SAMul
David K.
Smith

Department of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: david.smith@york.ac.uk
First published on 26th April 2018
Abstract
This feature article provides a personal insight into the research from my group over the past 10 years. In particular, the article explains how, inspired in 2005 by meeting my now-husband, Sam, who had cystic fibrosis, and who in 2011 went on to have a double lung transplant, I took an active decision to follow a more applied approach to some of our research, attempting to use fundamental supramolecular chemistry to address problems of medical interest. In particular, our strategy uses self-assembly to fabricate biologically-active nanosystems from simple low-molecular-weight building blocks. These systems can bind biological polyanions in highly competitive conditions, allowing us to approach applications in gene delivery and coagulation control. In the process, however, we have also developed new fundamental principles such as self-assembled multivalency (SAMul), temporary ‘on–off’ multivalency, and adaptive/shape-persistent multivalent binding. By targeting materials with applications in drug formulation and tissue engineering, we have discovered novel self-assembling low-molecular-weight hydrogelators based on the industrially-relevant dibenzylidenesorbitol framework and developed innovative approaches to spatially-resolved gels and functional multicomponent hybrid hydrogels. In this way, taking an application-led approach to research has also delivered significant academic value and conceptual advances. Furthermore, beginning to translate fundamental supramolecular chemistry into real-world applications, starts to demonstrate the power of this approach, and its potential to transform the world around us for the better.
Dave Smith is Professor of Chemistry at University of York, UK, publishing >150 research papers and with an h-index well over 50. He is a passionate educator with a strong belief in widening participation, giving outreach lectures to >50 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 UK school students and developing his own YouTube chemistry channel, with >500
000 UK school students and developing his own YouTube chemistry channel, with >500Beginnings
In 2006, I published a feature article in Chemical Communications.1 By that point, I had been an independent academic in York for 7 years and had developed a research programme exploring fundamental supramolecular chemistry. This built on my scientific training with Prof. Paul Beer at University of Oxford and Prof. Francois Diederich at ETH Zurich, which gave me a love of understanding how molecules interact with one another. Interactions between molecules can be considered analogous with interactions between people that have always fascinated me. I love to teach and do outreach, probably inspired by a long line of teachers in my family, all the way back to my Great Grandmother. Furthermore, my own education at a comprehensive school in Stockport demonstrated the value of those teachers that were able to effectively marshal their inter-personal interactions – exactly like the molecules we now try to manipulate. Just before writing that feature article 12 years ago, I made my own most important inter-personal interaction, with my husband-to-be, Sam. In many cases, as a research chemist, this would not matter, but in my case, this single event led to a radical change in how I approached research. This article aims to tell that story.Sam has cystic fibrosis – the genetic disease that encodes faults in chloride transport proteins within the cell membrane, which in turn causes mucus build-up around cells.2 This problem is particularly acute in the lungs, where the mucus hosts bacterial infections that ultimately lead to severe lung damage and early death. When I met Sam, my research team was pioneering an understanding of self-assembled gels in organic solvents3 and anion recognition events in non-aqueous media.4 I was, and indeed still am, very proud of this research, but explaining to someone with a life-limiting condition that you are using all of your skills to understand how peptides interact with one another in toluene somehow felt inadequate. I therefore became determined that some of our research would approach problems that might make a difference, specifically in CF-related medicine. In terms of supramolecular chemistry, this defines a clear challenge: for research to be medicinally or biologically relevant, you have to work in highly competitive media – for example water, and more likely serum, plasma, or even blood. This makes supramolecular nanomedicine highly challenging.5 We therefore speculated that by targeting medicinal applications, important new fundamental insights and guiding principles may also emerge.
From DNA binding to gene delivery
Perhaps the most widely discussed potential CF treatment is gene therapy – delivering a ‘healthy’ copy of the faulty gene to cells, where it can subsequently generate the required protein.6 Such an approach could treat cystic fibrosis at source, rather than just alleviating symptoms, as most current therapies aim to do. DNA is a polyanion, and I had expertise in binding anions dating back to my own PhD,7 so this therefore seemed to be a way of applying my knowledge to a problem of interest. Considering my own background in dendrimer chemistry,8 and the well-known capacity of dendritic molecules to achieve gene delivery,9 I had previously considered this area of research, but had rejected it as being too competitive. Now I had a real reason to join the competition.We decided to develop an innovative biomimetic approach to DNA binding and delivery. Given the well-known ability of multivalent displays of ligands to enhance binding affinities,10 we designed multivalent arrays of bio-inspired DNA-binding ligands. An effective naturally-occurring DNA binder is spermine,11 present in sperm. This compound was first isolated in crystalline form from human semen by van Leeuwenhoek in the late 17th century,12 testimony to its very high concentrations in seminal fluid. Spermine is also present at micromolar levels in all eukaryotic cells. It has pKa values that are optimised for DNA binding, is present as a tetra-cation at physiological pH, and is an appropriate shape to bind within the minor groove of DNA. However, the binding of an individual spermine unit to DNA has relatively low affinity.13 For this reason, relatively large concentrations are required in vivo to achieve control over DNA.
We reasoned that displaying spermine ligands in a multivalent manner may enhance the binding affinity, and our initial design therefore simply displayed these ligands on the periphery of dendritic architectures (G1-SPM and G2-SPM, Fig. 1A).14 Nona-valent G2-SPM was a highly effective DNA binder, with binding affinities effectively independent of salt concentration (i.e., ionic strength). Conversely, the binding of trivalent G1-SPM to DNA was weaker, and was adversely affected by physiological salt concentrations. This intriguing phenomenon was challenging to fully rationalise. At around this time, I joined a highly productive COST network (TD0802) focussing on Dendrimers in Biomedicine.15 Indeed, EU networking has been a huge part of the last 12 years of research. As a part of this network, I met the irrepressibly energetic Prof. Sabrina Pricl from University of Trieste, who persuaded me multiscale modelling was a vital complement to our experimental work. This in silico approach can help understand binding mechanisms and design agents ab initio with greater activity. Sabrina has become a close collaborator and a great personal friend. In 2009, in a key paper, we proposed the underlying mechanism by which G2-SPM resists competitive salt ions at the binding interface – a ‘screening and sacrifice’ model.16 Not all of the nine spermine ligands actively bind to DNA and in high salt conditions, more of the ligands sacrificed their interactions, but then acted to screen the remaining binding sites, which achieved significantly enhanced affinity (Fig. 1B). We therefore demonstrated an important new advantage of having flexible multivalent arrays under these highly competitive binding conditions (Fig. 1C). Pleasingly, this demonstrated that wholly new fundamental insights can arise from work that starts out with a specific application in mind.
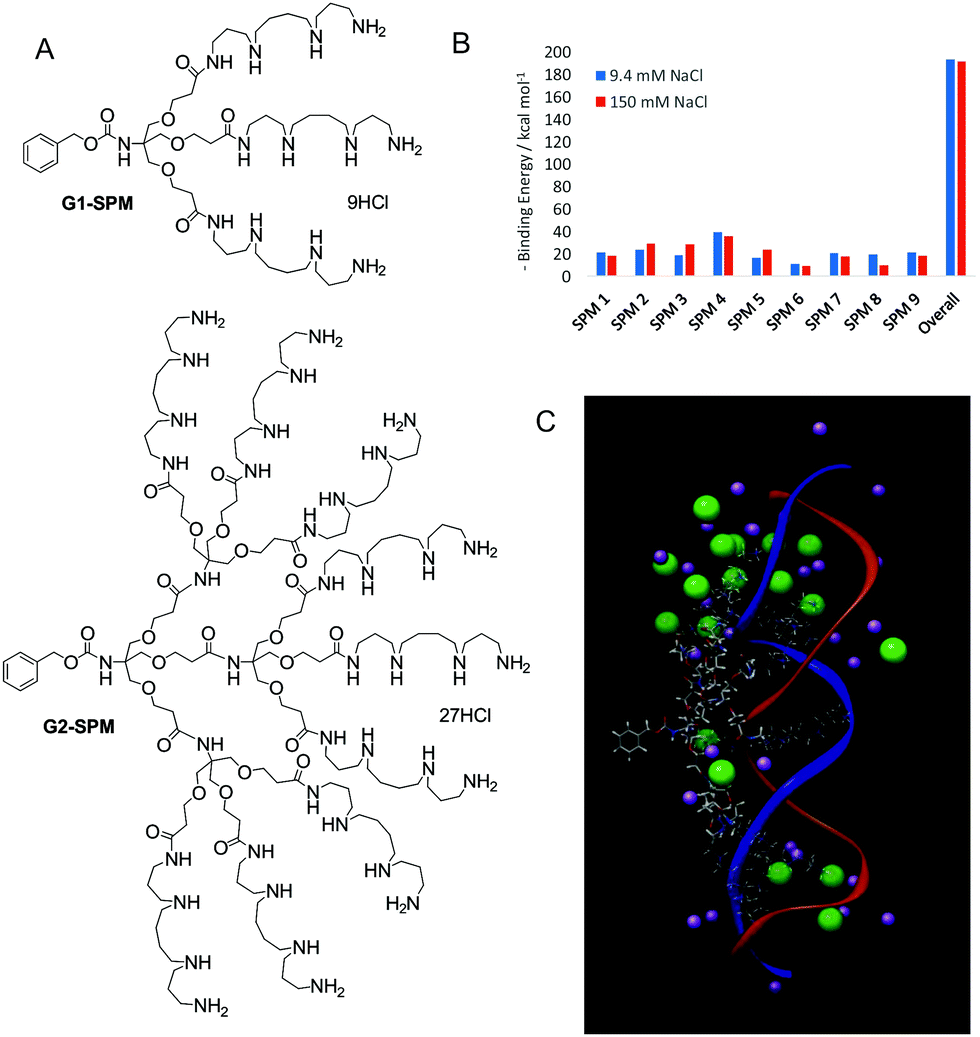 | ||
| Fig. 1 (A) Dendrons G1-SPM and G2-SPM designed as multivalent DNA binders. (B) Graph derived from molecular dynamics indicating that on addition of high concentrations of salt the overall binding is hardly affected – a result of several ligands (SPM6 and SPM8) sacrificing all interaction with DNA while other ligands (SPM2 and SPM5) then optimise their binding via shielding. (C) Image from molecular dynamics (MD) simulation of G2-SPM (skeletal structure) bound to DNA (blue and red ribbons) in the presence of NaCl (purple/green). | ||
Given the strength of interactions between G2-SPM and DNA, we recognised that degradation of our DNA binders would be important if we were to achieve effective gene delivery. Our synthetic approach was therefore modified to change Newkome-type amide–ether dendritic scaffolds,17 into Fréchet–Hult type ester-derived dendrons (G1-Ester, G2-Ester and G3-Ester, Fig. 2, top).18 Fréchet–Hult dendrons had been reported to degrade under biologically-relevant conditions.19 Displaying our DNA binding ligands on the surface of such systems yielded dendrons that exhibited ‘on–off’ multivalency.20 This was a new concept, developed by us, in which the synthetic construct initially has ultra-high-affinity for its target owing to its multivalent structure, but after degradation, the multivalent array is destroyed, and all ability of the compound to intervene in biological pathways is lost. We reasoned this approach would assist with intracellular DNA release and limit the persistence and potential toxicity of our nanoscale delivery vehicles – a problem often associated with dendrimers and other nanosystems in vivo.21 We also developed dendrons in which a photo-cleavable linker was inserted between the dendron and the ligand display, so that UV irradiation led to triggered loss of the ligands from the surface of the dendron (Fig. 2, bottom) hence switching off the ultra-high affinity DNA binding event.22
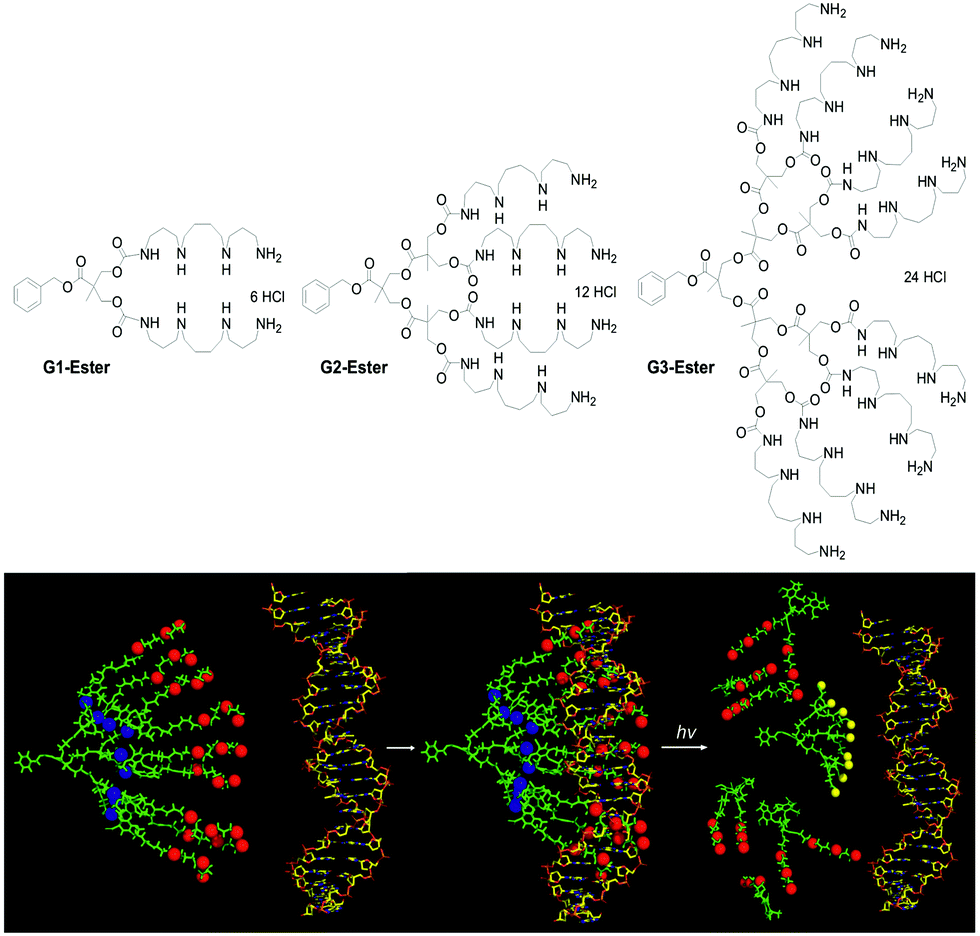 | ||
| Fig. 2 (top) Structure of degradable DNA binding dendrons capable of ‘on–off’ multivalency based on the Fréchet–Hult scaffold, and (bottom) schematic of UV-degradable dendrons including a photo-cleavable linker between the dendron and the SPM ligands, indicating cleavage of dendron and loss of DNA binding ability. | ||
In addition to tuning the framework of the dendron to introduce degradability, we also tuned the ligands on the surface (e.g.G1-SPM, G1-DAPMA, G1-DAP, Fig. 3).23 We found that naturally occurring SPM ligands were the most effective for DNA binding, but that less effective DAPMA ligands actually gave somewhat better gene delivery profiles. Molecular dynamics was insightful in understanding the thermodynamic impact of ligand modification. Although the enthalpic contribution to binding was roughly proportional to the dendron surface charge, dendrons with DAPMA surface amines had significant entropic costs of binding to DNA. This is a consequence of the fact that for this diamine more of the dendron structure has to be organised in order for multiple charges to make effective contact with DNA, while for SPM, each surface ligand is already an optimised triamine, giving each individual charge a lower entropic cost of binding. Furthermore, the hindered tertiary amine in DAPMA bound particularly strongly to the DNA double helix leading to ligand back-folding and geometric distortion of the DNA. Although this weakens binding, we suggested it might explain enhanced gene delivery, as DNA compaction and release are both important steps.
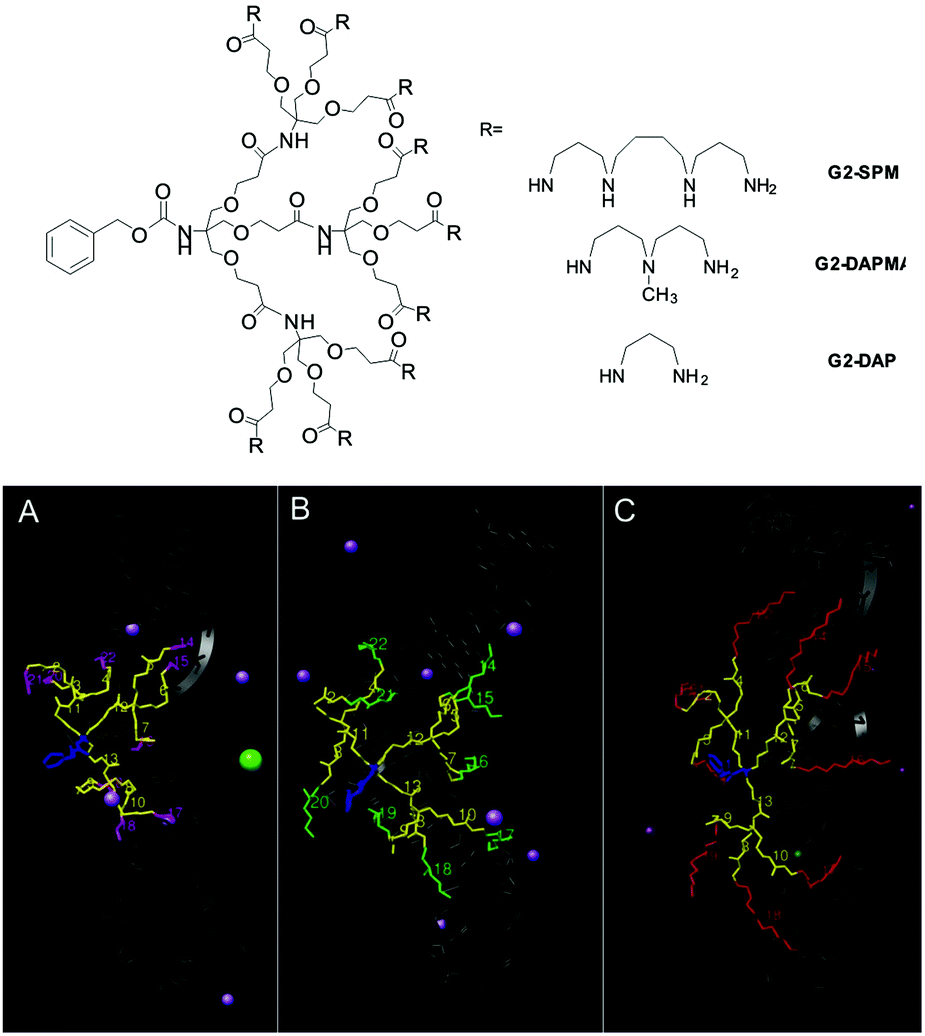 | ||
| Fig. 3 (top) Structures of dendrons with different ligands and (bottom) snapshots of molecular dynamics simulation of dendrons binding to double helical DNA: (A) G2-DAP, (B) G2-DAPMA and (C) G2-SPM. Within the dendron CEN is shown in blue, REP in yellow and the amine ligands are shown in magenta (DAP), green (DAPMA) and red (SPM). The DNA is portrayed as a dark gray shadow, water molecules are omitted for clarity, and only those counterions in close proximity to the complexes are shown. | ||
From multivalency to self-assembled multivalency (SAMul)
All of the systems described above, however, had significant synthetic complexity, and relied on relatively elaborate covalent synthesis to create multivalent ligand displays. We therefore wondered if instead of using covalent synthesis to form higher generation multivalent dendrons, we could use a self-assembly step to multiply up smaller low-affinity binding arrays into larger high-affinity nanostructures. This approach is intermediate between the two main approaches to non-viral gene delivery vectors, which rely on either cationic polymers24 or cationic lipids.25 We reasoned that our systems may combine some of the advantages of each of these different classes of vector.Initially, we simply modified the focal point of our existing dendrons with hydrophobic units (e.g.Chol-G1, Chol2-G1, and Chol-G2, Fig. 4).26 Pleasingly, converting a simple benzyloxycarbonyl protecting group at the focal point of our small first generation dendrons into a cholesterol unit, to make Chol-G1 switched on ultra-high affinity binding of DNA. Furthermore, although Chol-G2 had a much larger binding array than Chol-G1 it was actually much less effective (i.e. less is more).27 Multiscale modelling studies indicated that Chol-G2 is unable to self-assemble effectively as it is dominated by the hydrophilic, large dendritic head group – the poorly packed SAMul nanostructure therefore had a much lower surface charge density (Fig. 4B). This demonstrates the importance of balancing the potential of self-assembly and multivalency to optimise binding. We were not the first in the literature to recognise that self-assembly could organise multivalent ligand displays – indeed, this was reported by the Whitesides group as early as 1992,28 but we did bring this disparate field together in a key review of the quantitative studies of self-assembled multivalency.29 We went on to refer to self-assembled multivalency as ‘SAMul’, in honour of my husband, Sam (Samuel) Smith.
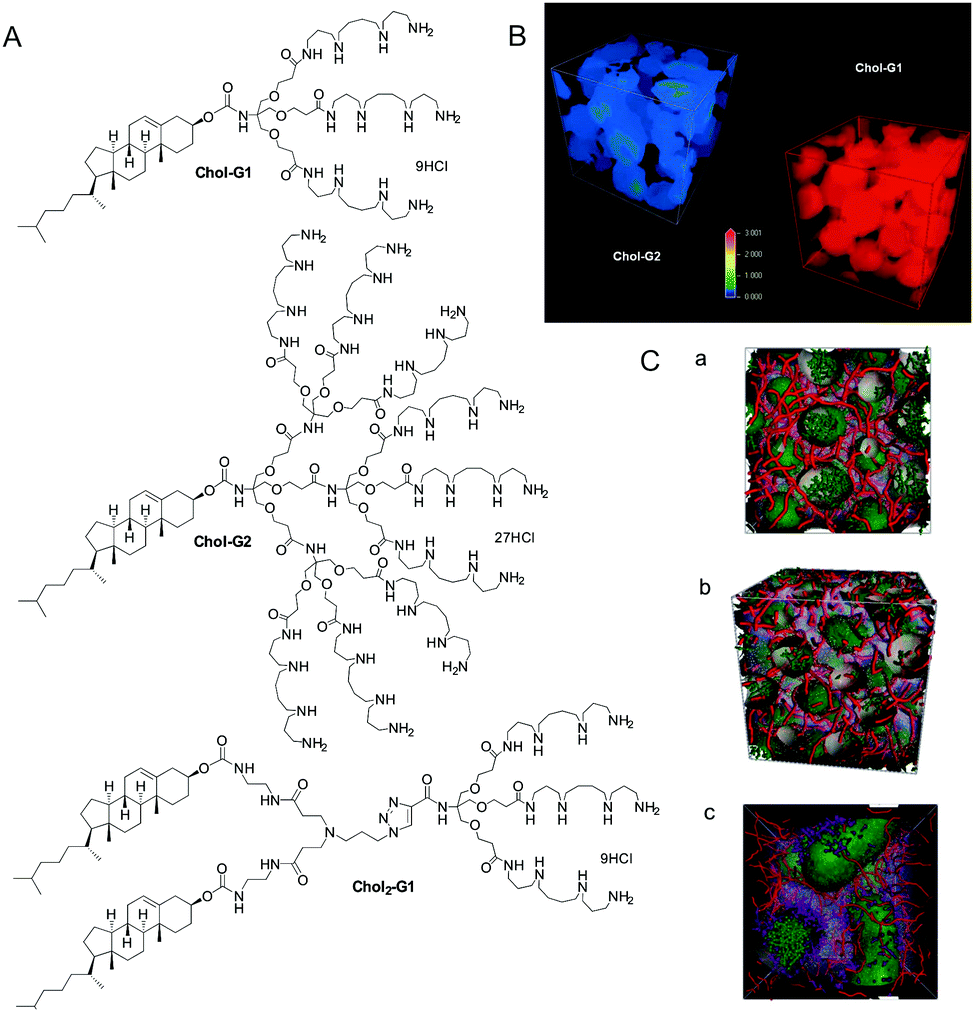 | ||
| Fig. 4 (A) Systems developed for the self-assembled multivalent (SAMul) approach to DNA binding. (B) Mesoscale modelling demonstrating that Chol-G1 actually has a higher cationic surface charge than Chol-G2 as a result of its more effective assembly – ‘less is more’. (C) Mesoscale modelling at elevated concentrations of self assembled Chol-G1 (a), Chol-G2 (b) and Chol2-G1 (c) shown in green binding to DNA shown in red. Note the different cylindrical morphology preferred by Chol2-G1. | ||
It is tempting to think that a SAMul binding effect will either be turned ‘on’ or ‘off’ depending simply on whether or not the system is, or is not, self-assembled. However, in a structure–activity relationship study we varied the hydrophobe and found that this simple relationship was not the case.30 Hydrophobes that were better able to drive self-assembly gave rise to self-assembled micellar nanostructures with higher surface charge densities. Indeed the cationic charge density could be tuned across a range. These systems then showed tunable DNA binding affinities primarily as a result of their different charge densities, resulting from the hydrophobic tuning. This clearly demonstrates that SAMul systems can have tunable binding affinities depending on the precise structure.
When modified with hydrophobic units, the gene delivery potential was also enhanced26 over the baseline results for the non-self-assembling dendrons.31 We reasoned that the cationic lipid nature of these systems assists delivery – it is known that lipids can enable endosomal escape, via a flip-flop mechanism.32 The best vector was Chol2-G1, which has a relatively large hydrophobic component. Multiscale modelling suggested this system could assemble into cylindrical micelles at elevated concentrations typical of, for example, endosomes, and form a hexagonal phase.27 This is in accord with Israelachvili principles of self-assembly that state that the hydrophobic/hydrophilic ratio of lipids program-in the preferred assembly mode of surfactants.33 It is known that lipids capable of forming hexagonal phases are preferred for endosomal escape.34 We also found there were synergistic advantages for gene delivery of mixing lipid-like and polymer-like systems.
We explored related morphological effects on the self-assembly of multivalent systems using a series of RGD-peptide modified lipids. RGD peptides can enhance cell targeting owing to interactions with integrin receptors on cell surfaces35 – we developed such systems to mix into our gene delivery vehicles and enhance delivery. We initially demonstrated that self-assembled RGD-peptides bound integrins as effectively as covalent dendritic multivalent equivalents – a clear SAMul effect.36 Once again, the hydrophobic/hydrophilic balance modified the morphology of the SAMul systems.37 Spherical assemblies performed better in our integrin binding assay than cylindrical micelles, possibly because of the more effective ligand display, with each head group having more space at the highly curved spherical surface, therefore demonstrating that SAMul binding responds to nanoscale morphology.
In a key study, we brought together our new concepts, and reported a family of degradable SAMul dendrons (e.g.Chol-G2Ester-DAPMA, Chol2-G2Ester-DAPMA and C22-G2Ester-DAPMA, Fig. 5) and explored their DNA binding and gene delivery potential.38 Although these compounds showed useful degradation profiles under physiological conditions of pH and ionic strength, they did not degrade once bound to DNA. We determined that hydrolysis of the esters within the structure was accelerated by the peripheral amine ligands via an intramolecular catalytic acceleration at physiological pH (Fig. 5B). Hydrolysis was switched off at lower pH values (owing to amine protonation) and in the presence of DNA (owing to amine–DNA binding). Sadly, this degradation profile was therefore less useful for gene delivery in vitro and DNA release is problematic. As such, these results were somewhat disappointing – although they led to significant breakthroughs in other potential areas of therapy, as described later.
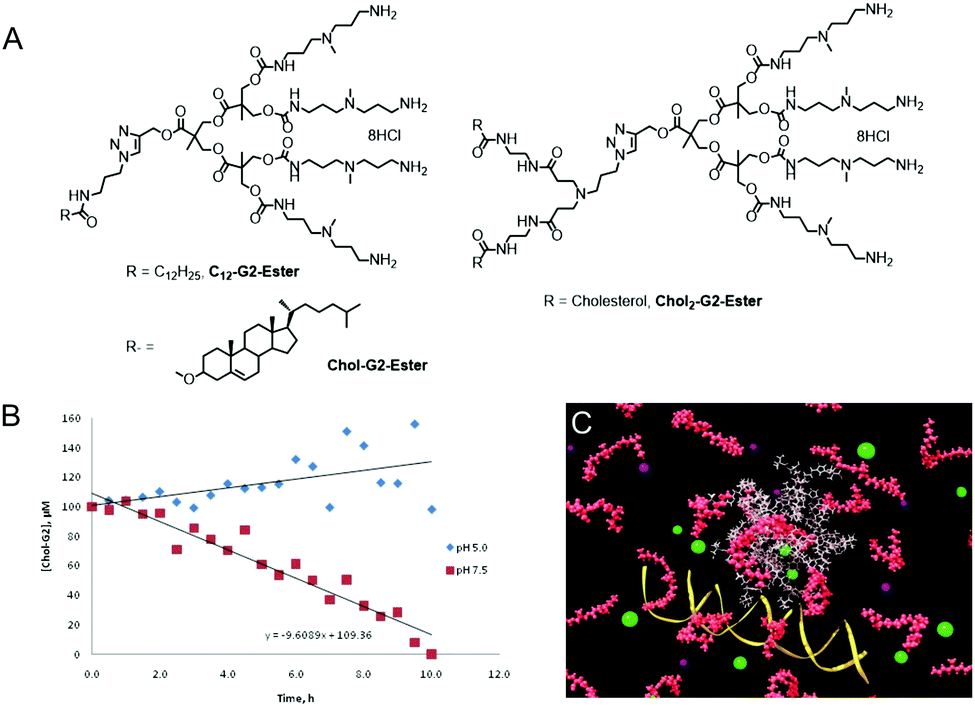 | ||
| Fig. 5 (A) Structures of SAMul degradable dendrons. (B) Degradation of dendron at pH 7.5 with stability at pH 5.0. (C) Computational modelling of degradation and disassembly of SAMul dendron array demonstrating how in principle, DNA release should be achieved. | ||
In addition, we co-assembled PEG lipids with a SAMul dendron to modify the DNA binding affinity.39 One of these modified the ability of the co-assembly to cross a model mucus layer, an important step in therapeutic delivery to the CF lung. We rationalised these effects in terms of a balance between the mucoadhesivity due to the surface charge of the nanoscale aggregates and that due to the PEG groups. This clearly demonstrated the potential of co-assembly as a simple strategy for modifying and optimising the performance of SAMul systems for biomedical use.
We also reported a ‘double degradable’ approach to SAMul systems in which a disulfide linker was incorporated between the hydrophobic unit and the multivalent head group (Chol-SS-G2-Ester, Fig. 6).40 This allows these structures to undergo triggered reductive cleavage, with dithiothreitol (DTT) inducing controlled breakdown, enabling the release of bound DNA – another example of temporary ‘on–off’ multivalency. Furthermore, because the multivalent dendrons are constructed from esters, a second slow degradation step takes place converting the dendron into very small molecular-scale building blocks, clearly illustrating the potential of such self-assembled systems including multiple degradable units.
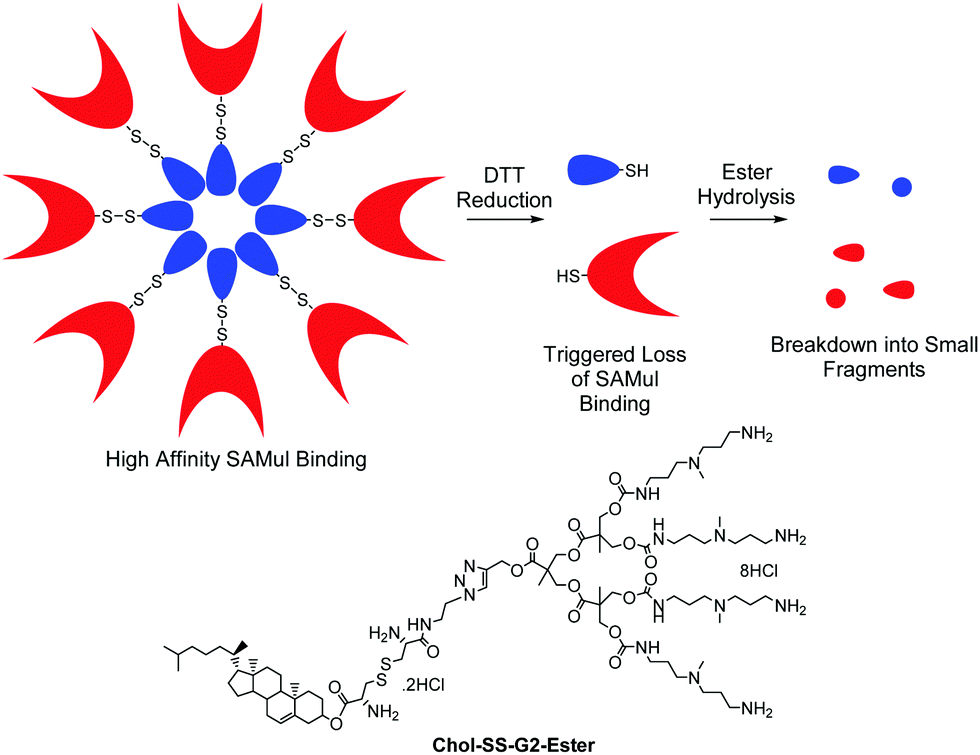 | ||
| Fig. 6 Double degradable SAMul DNA binding arrays which disassemble as a result of disulfide reduction and ester hydrolysis and the structure of Chol-SS-G2-Ester which possesses two different types of degradable unit. | ||
To further translate our approach towards the clinic, in collaboration with Rainer Haag, Marcelo Calderon and co-workers in Berlin, we developed SAMul systems using the general principles described above but with modified molecular designs. For example, our SAMul concept was combined with UV-degradable linkers to generated SAMul systems that were switched off by UV irradiation.41 We also developed other systems (e.g., G2-Ether (A1), Fig. 7) and tested them for siRNA delivery both in vitro and in vivo.42 The ester was in a different orientation to our previous design, and further from the focal point of the dendron, which was active for siRNA delivery, effectively achieving gene silencing in the tumour cell line 786-O (Fig. 7B). It was reasoned that the ester had an effective degradation and siRNA release pathway. Pleasingly, in vivo studies demonstrated that these delivery vehicles, when combined with siRNA, did not elicit major pro-inflammatory cytokine response when tested in vivo in mice (Fig. 7C). Such responses are a frequent problem with gene delivery vehicles,43 and other nanoscale therapeutics, and their absence is very desirable. We anticipate such systems will go on to further development.
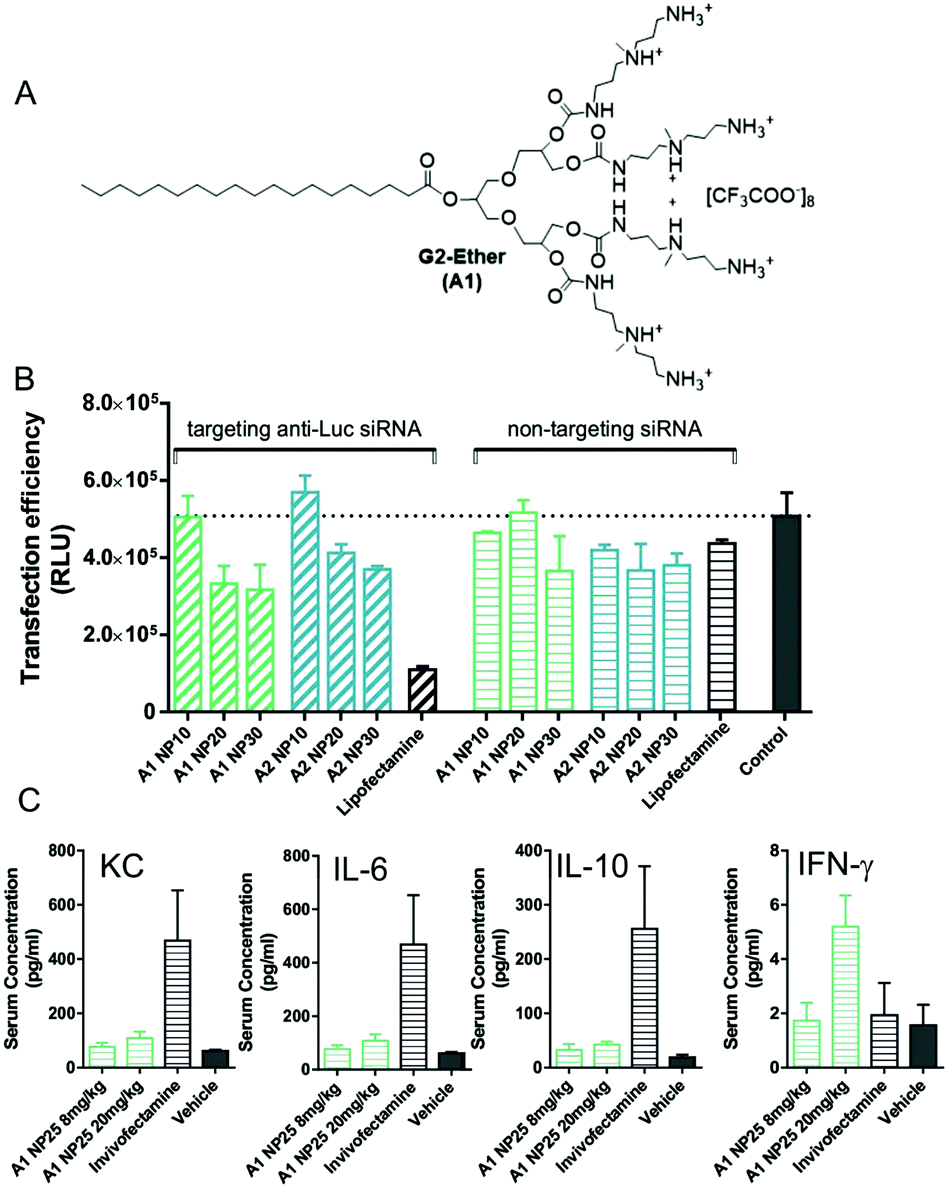 | ||
| Fig. 7 (A) Structure of dendron developed in collaboration with Haag and co-workers. (B) 786-O Luc transgenic cells were transfected with luciferase specific and non-targeting siRNA at N/P ratios of 10, 20, and 30 for 48 h. Lipofectamine was used as a positive control and untreated cells as the negative control. (C) In vivo study of mice treated intravenously with 8 mg kg−1 and 20 mg kg−1 complexed with non-targeting siRNA at N/P ratios of 25, or Invivofectamine. Retrobulbar blood was taken 1 h after injection and serum was examined via Meso Scale Discovery Multi-Spot Assay System, Mouse ProInflammatory 7-Plex Assay Ultra-Sensitive Kit. Results are shown as mean ± SD of triplicates. | ||
We are also delighted that other researchers have applied our SAMul methodology with a high degree of success to siRNA delivery. Notably Ling Peng and her research team developed self-assembling dendritic systems that showed high levels of activity in vitro and considerable potential in vivo.44 We are also now working with researchers across the EU to try and translate some of our gene delivery vectors further, with the ultimate goal of making an impact on cystic fibrosis healthcare. We hope that, in one way or another, enhanced treatments may be forthcoming from key concepts developed here in York, such as SAMul binding and temporary (‘on–off’) multivalency.
How major surgery inspired SAMul heparin binders and sensors
In 2010, around the time of our civil partnership, Sam's health rapidly deteriorated, and his lung function dropped to just 20%. For cystic fibrosis patients with heavily progressed disease like this, the only clinical option is lung transplantation. Sam was listed for transplantation and was fortunate enough to receive a transplant at the Freeman Hospital (Newcastle) in January 2011. Obviously, this was a hugely stressful period in both of our lives. However, once again, it gave me plenty of opportunity to reflect on the chemistry we were doing and other uses it may find. On discussion with surgeons and anaesthetists, I realised that a key drug during surgery was heparin – a polyanionic saccharide that acts on the thrombin cascade preventing undesired coagulation and clotting.45 Once surgery is complete, and the patient is returned to the ward, it is important to achieve homeostasis so that clotting and recovery can begin. This is achieved using a heparin-binding agent, protamine, which removes heparin from the bloodstream, enabling clotting. Protamine is a polycationic protein, which binds heparin via electrostatic ion–ion interactions using its multiple arginine residues.46 However, a significant number of patients have adverse responses to protamine,47 meaning it cannot be used in high doses, giving rise to problems with ‘heparin rebound’,48 when heparin desorbs from plasma proteins some hours later leading to bleeding. I therefore learned there was significant interest both in binding heparin (to remove it once surgery is complete) and sensing heparin (to determine precisely how much is present in vivo). There have been a number of synthetic approaches to both heparin binding49 and heparin sensing,50 and we have reviewed this area of translational supramolecular chemistry.51While I was sat at Sam's bedside, I reflected on the fact that some of our gene delivery agents, which bind polyanions as described above, suffered from the problem of degrading in the absence of DNA, but not when bound to it.38 Although problematic for gene transfection, this is potentially useful for heparin binding – indeed, the ideal heparin rescue agent should be stable when bound to heparin, so excretion of the intact complex can occur, but should degrade if present in excess. This would potentially allow the patient to be dosed with relatively large amounts of the active agent, as unbound material would degrade into non-toxic by-products. In the case of self-assembled multivalent (SAMul) systems, degradation will also switch off high-affinity binding and thus prevent intervention in other biological pathways.
We therefore developed a simple SAMul system C22-G1 (Fig. 8) that could bind heparin with high affinity.52 This system self-assembled into nanostructures at low concentration (4 μM), displaying cationic ligands capable of heparin binding. The linker between ligands and hydrophobe contained an ester, capable of degrading under physiological conditions. When dried, C22-G1 nanostructures were surprisingly robust, and even in the presence of strongly binding heparin, transmission electron microscopy (TEM) indicated they did not change their spherical nanoscale morphologies, suggesting good stability. On drying with heparin, the micelles organised around the polyanionic heparin to form hierarchical aggregates. More recent studies in collaboration with Mauri Kostiainen have fully characterised the organisation of cationic spherical micelles with polyanionic heparin using small angle X-ray scattering and multiscale modelling.53 This is hierarchical electrostatic self-assembly, in which spherical cationic assemblies close-pack with cylindrical anionic polymers to give face-centred-cubic arrays ca. 50–250 nm in size.
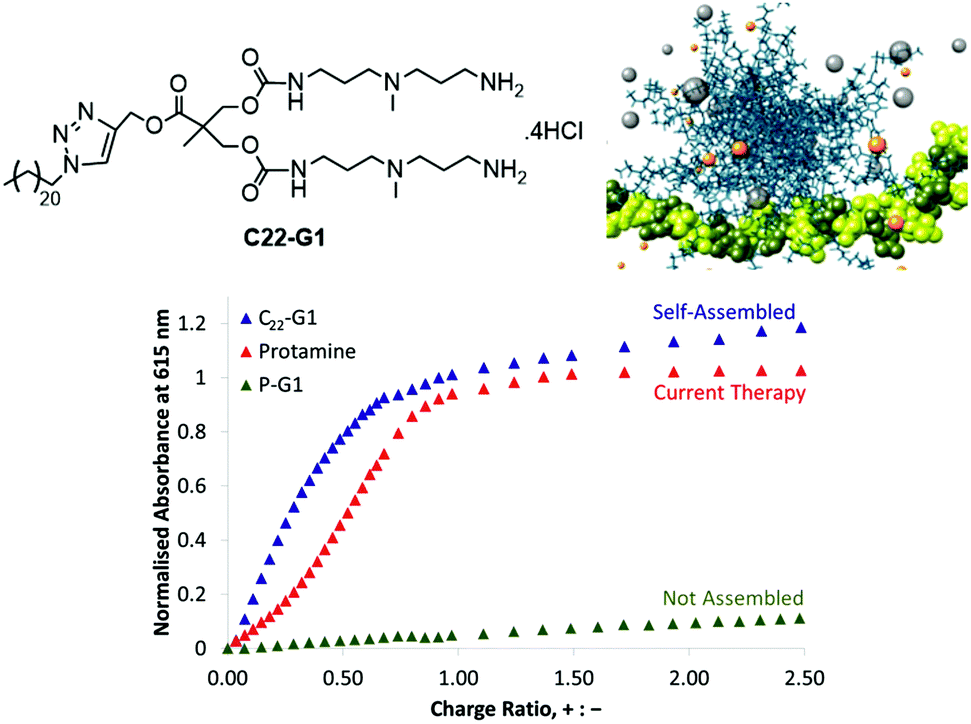 | ||
| Fig. 8 Structure of C22-G1, multiscale modelling simulation of C22-G1 self-assembled into a multivalent structure and bound to heparin, and binding study comparing the ability of C22-G1, current therapy protamine, and a non-self-assembling precursor (P-G1) indicating that in buffer, the self-assembling nanosystem outperforms protamine, and that there is a clear quantifiable SAMul binding effect. | ||
In a later study, we fully characterised C22-G1 nanostructures in solution using dynamic light scattering (DLS). and quantified the binding ability of C22-G1 in more detail. In buffer, C22-G1 was a more effective heparin binder than protamine (Fig. 8).54 Furthermore, comparison of the heparin binding of C22-G1 with an analogue with no hydrophobic unit demonstrated that the C22 chain switches on high-affinity binding – a clear SAMul effect. However, the presence of serum somewhat disrupted the ability of C22-G1 to self-assemble and hence bind heparin – we reasoned this was due to the C22 unit interacting with serum albumin proteins. This can be a problem with self-assembled systems, which can be kinetically labile, and hence disassemble in the presence of competitive influences.55 Nonetheless, this lead compound was active in the PT anti-coagulation assay in human plasma, reversing the anti-coagulant effect of heparin when applied at a clinically useful dose – demonstrating that the SAMul approach can operate in highly competitive, biologically relevant media. We have further stabilised our heparin binders in competitive conditions and developed unpublished modified lead compounds for potential in vivo study.
We have reported the impact of self-assembled morphology on heparin binding using (C12)2-Lys and (C12)2-Lys-Lys2 (Fig. 9).56 As a result of their hydrophobic/hydrophilic ratios, the former compound assembles into worm-like micelles while the latter forms spherical micelles. Although the spherical micelles bind heparin more effectively in buffer because of their more open surfaces and better ligand display, the cylindrical micelles have greater stability against competitive influences, and better maintain their binding ability when buffer is replaced with human serum.
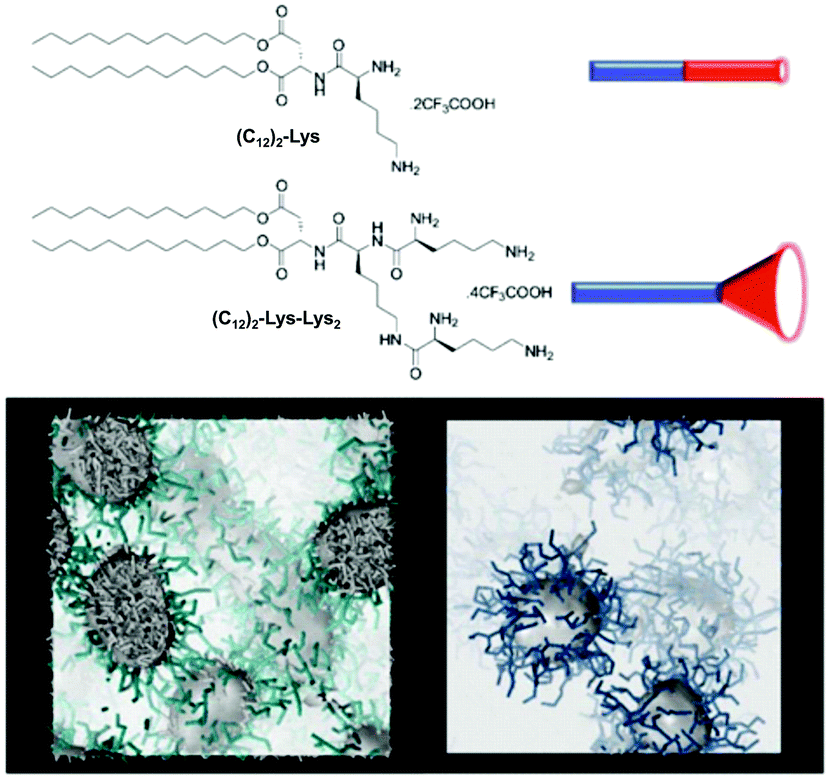 | ||
| Fig. 9 Compounds (C12)2-Lys and (C12)2-Lys-Lys2 have different hydrophobic/hydrophilic ratios and different molecular shapes, which give rise to self-assembled nanostructures with different morphologies – cylindrical micelles and spherical micelles respectively – as indicated by multiscale modelling. | ||
We have also developed heparin sensors. In part, we wanted a heparin sensor for rapid quantification of the relative abilities of our heparin binders via a dye displacement assay. However, there is also a clinical need for heparin sensors that could rapidly assess the total heparin load in a patient's bloodstream, allowing anaesthetists to accurately provide a patient with additional heparin, or heparin-rescue agent. At present, heparin sensing is achieved by performing coagulation assays on blood samples – a relatively time-consuming, off-line process – clearly there is scope to improve this. Amongst heparin sensors in the literature, Azure A was reported to work in highly competitive conditions, and should have been ideal for our purposes.57 However, we were unable to reproduce this, and noted the original experiments were not buffered and may have suffered from pH changes. We therefore decided to develop a new dye with higher affinity towards heparin by modifying the well-known dye thionine acetate with cationic arginine units.58 This was achieved using a simple two-step methodology based on peptide coupling and deprotection in a pleasing overall yield (Fig. 10). We named the resulting dye ‘Mallard Blue’ (MalB) after the steam train of the same colour, which holds the world speed record and is housed in the National Railway Museum here in York (ever since I was a boy I have always loved trains!). This dye is an effective heparin sensor in highly competitive media, and can rapidly detect heparin at clinically relevant concentrations in human serum.
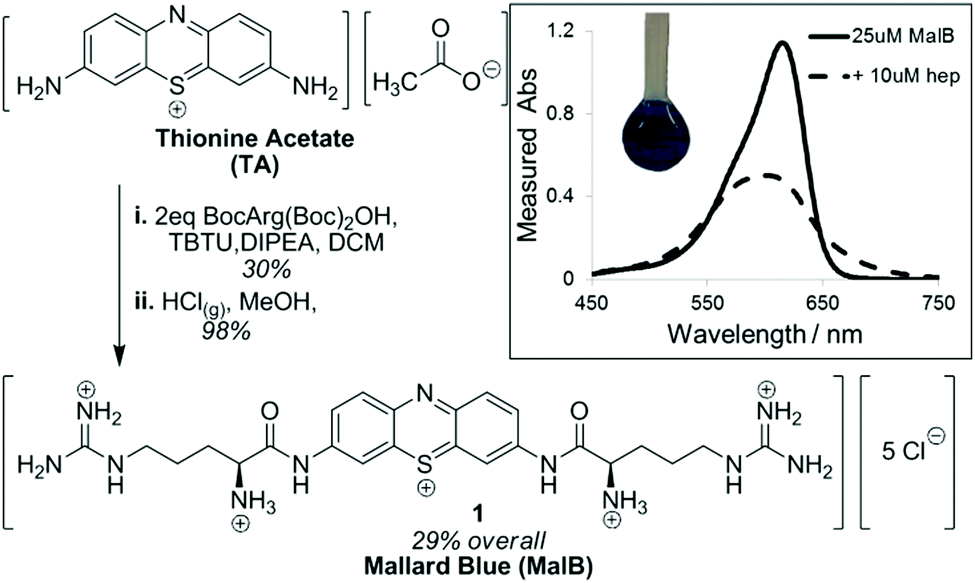 | ||
| Fig. 10 Synthesis of Mallard Blue based on modification of thionine acetate with Boc-protected arginine, followed by Boc deprotection. (inset) UV-vis response of MalB to the addition of heparin. | ||
Using MalB, we developed a dye displacement assay for quantification of the performance of heparin binders.59 To demonstrate this assay in action, a family of dendritic compounds with the capacity to bind heparin was investigated – flexible PAMAM dendrimers and more rigid ‘Transgeden’ (TGD) dendrimers (which have peripheral PAMAM units attached to a conjugated core).60 This study indicated fundamental differences between these two families of dendrimers. In the presence of excess heparin, a clinically relevant situation at the start of heparin rescue, TGD outperforms PAMAM because the rigidity of TGD means small clusters/patches of surface amines are locally well-organised for binding. However, when less heparin is present (towards the end of heparin rescue), the overall rigid nanoscale structure of TGD is not sufficiently flexible to maximise all of its contacts with a single heparin binding partner (Fig. 11). We described TGD as showing shape-persistent multivalency. Conversely, in PAMAM dendrimers, the individual surface ligands are not so well locally optimised for target binding, but the flexibility of the overall nanostructure allows it to adapt its global shape more easily and hence maximise the total number and efficiency of contacts with the binding partner (Fig. 11). We referred to this behaviour as adaptive multivalency. We suggest that these fundamental differences are of considerable significance in multivalent binding. Once again therefore, adventures into applied research gave rise to new insights.
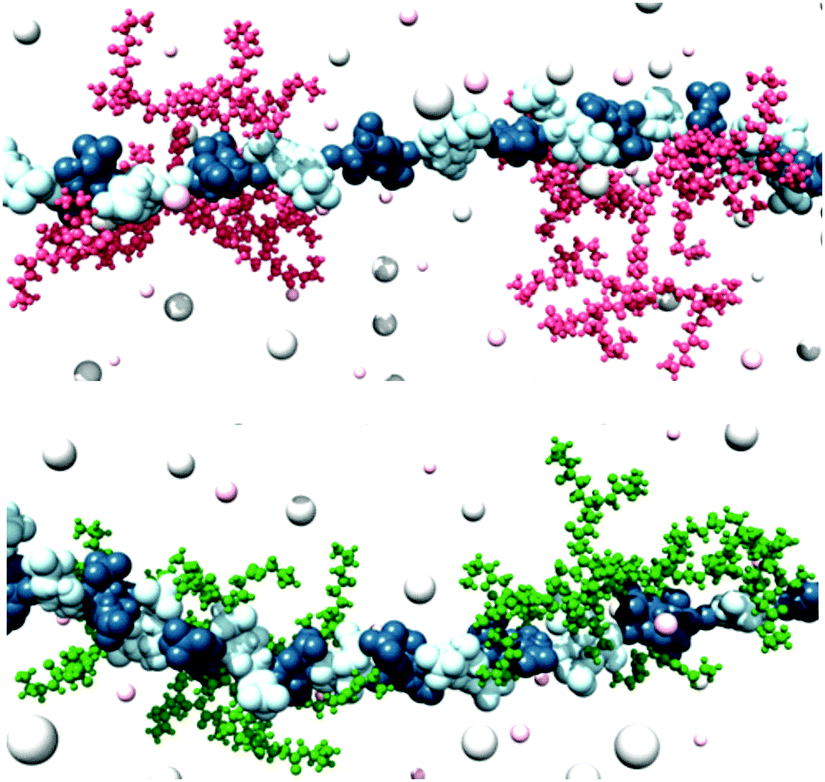 | ||
| Fig. 11 (top) TGD-G2 and (bottom) PAMAM-G2 dendrimers bound to heparin at a charge excess of 0.4 using MD modelling methods. TGD and PAMAM molecules are portrayed as pink and green sticks-and-balls, respectively. Heparin is shown as a chain of L-iduronic acid (blue) and D-glucosamine (light blue) alternating units. Some Na+ and Cl− counterions are shown as small and large pink and grey shaded spheres respectively. Water is not shown for clarity. It is evident that PAMAM is better able to adapt its structure and bind along the polyanionic heparin chain, unlike shape-persistent TGD. | ||
To develop heparin sensor technology further, it was desirable to develop sensors with an easily detectable fluorescent output, at two different wavelengths for ratiometric sensing. We therefore developed a heparin sensor incorporating pyrene.61 In buffered water, the sensor formed self-assembled multivalent (SAMul) nanostructures providing it with a significant (order of magnitude) improvement in dynamic range. Further, it gave a naked eye response unlike other pyrene derivatives. At elevated heparin concentrations, SAMul-enhanced sensing was also observed even in 100% serum. However, SAMul-enhanced sensing did not persist in serum at biomedically relevant concentrations. It is therefore interesting to reflect just how good the heparin sensing capacity of Mallard Blue is, being able to detect the polyanion at very low concentrations even in highly competitive conditions. This dye must be particularly optimised for the structural motifs dominant in heparin. Indeed, these observations led us to reflect on the role structural optimisation plays in electrostatic binding.
Selectivity in electrostatic binding – towards the polyanion world
Once involved in electrostatic polyanion binding, perhaps the most commonly asked question, is the extent to which selectivity can be designed into such systems. Many a paper reviewer or conference attendee has noted that ‘any polycation can bind any polyanion’ – one adding that ‘even Monsieur Coulomb himself would not have been surprised by these results’. Obviously, there is some truth in this, and models of ion–ion binding suggest that charge density plays a controlling role with deviations from this being rare.62 Indeed, even biology can struggle to achieve selectivity between different polyanionic targets – for example, it is known that DNA and heparin can, in some cases, both bind to the same protein targets.63 However, biology is a polyanionic world – nucleic acids, cell membranes, glycosaminoglycans, proteoglycans, tubulins, etc. are all negatively-charged.64 Increasingly, there are hints that some selectivity can be achieved in the polyanion world – for example, consensus sequences have been discovered for heparin binding, which would suggest that certain motifs evolved for this purpose.65 Nonetheless, achieving any degree of selective recognition using synthetic systems would appear to be a challenging fundamental target and it captured our attention.We developed chiral SAMul systems that achieved enantioselective polyanion binding (Fig. 12).66C22-L-Lys and C22-D-Lys are identical in every way apart from the chirality of the cationic lysine ligands that end up displayed on the surface of the SAMul nanostructure. Obviously, these two enantiomeric ligands have identical critical micelle concentrations, and form systems with identical dimensions and surface charges – just different chiralities. Interestingly, however, they bound to heparin and DNA with significant differences. Heparin bound preferentially to C22-D-Lys, while DNA had a preference for C22-L-Lys. This clearly demonstrates that charge density is not the only factor controlling binding affinity, and that the precise molecular-scale optimisation of the nanoscale binding interface plays a key role. Although chiral recognition of these polyanions is not new,67 it was surprising that our simple SAMul nanostructures could achieve this. Furthermore, this was the first report in which different polyanions expressed opposite enantioselectivities. Given the crucial roles played by each of these highly charged anions, we speculate whether such selectivities may help biology regulate the ubiquitous and complex ‘polyanion world’.
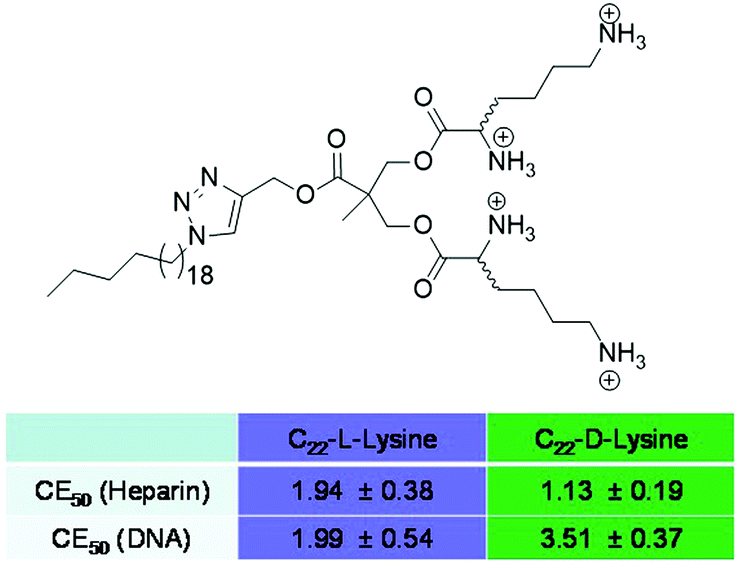 | ||
| Fig. 12 Compounds C22-L-Lys and C22-D-Lys and the charge excess of each required to achieve 50% displacement of MalB from heparin and EthBr from DNA at pH 7.4 (Tris–HCl, 10 mM, NaCl, 150 mM). Evidently, heparin binds more strongly to C22-D-Lys and DNA binds more strongly to C22-L-Lys. | ||
Extending this research further, chiral amino acid modifications were made to a very simple amphiphile (C16-Lys and C16-Gly-Lys).68 The presence of a glycine spacer unit between the hydrophobic unit and the cationic head-group was essential in enabling chiral recognition. Modelling proposed that the glycine spacer changed the polarity and shape of the amphiphile and hence modified the self-assembled morphology, enabling optimisation of the binding interface and yielding greater selectivity.
We also explored the impact of ligand modification on polyanion binding in SAMul systems.69 Palmitic acid was modified with a series of amines with different charges, yielding three different micellar assemblies (C16-SPM [4+], C16-SPD [3+] and C16-DAPMA [2+]) (Fig. 13). Overall, C16-SPM formed the loosest assembly, while C16-DAPMA was the most effectively packed with the highest surface charge. Intriguingly in our DNA-binding assays, DNA bound C16-SPM > C16-DAPMA > C16-SPD, whereas heparin bound C16-SPD > C16-SPM > C16-DAPMA. Clearly therefore, neither simple ligand charge nor micellar surface charge density, control the binding event. A combination of experimental (isothermal calorimetry, ITC) and simulation (multiscale modelling) techniques provided insight into the thermodynamics of binding. We proposed inherent differences between the polyanions, suggesting that DNA was shape-persistent while heparin was adaptive (Fig. 13). As such, shape-persistent DNA simply binds the charges it is presented with as best as it can. Spermine ligands are well adapted to bind DNA, have the highest individual charge, and therefore C16-SPM was the preferred binding partner. Conversely, adaptive heparin can modify its shape on binding to the SAMul nanostructure, meaning both ligand and polyanion optimise their mutual interactions. This led to C16-SPD being preferred, with heparin adapting and optimising each individual electrostatic interaction. This work indicates how selectivity may be achieved in the polyanion world, and suggests fundamental differences between polyanions. Interestingly, flexibility has been highlighted as a key beneficial factor in biological heparin binding proteins,70 which would agree with this view of heparin as an adaptive binding partner and suggest it might have genuine biological significance.
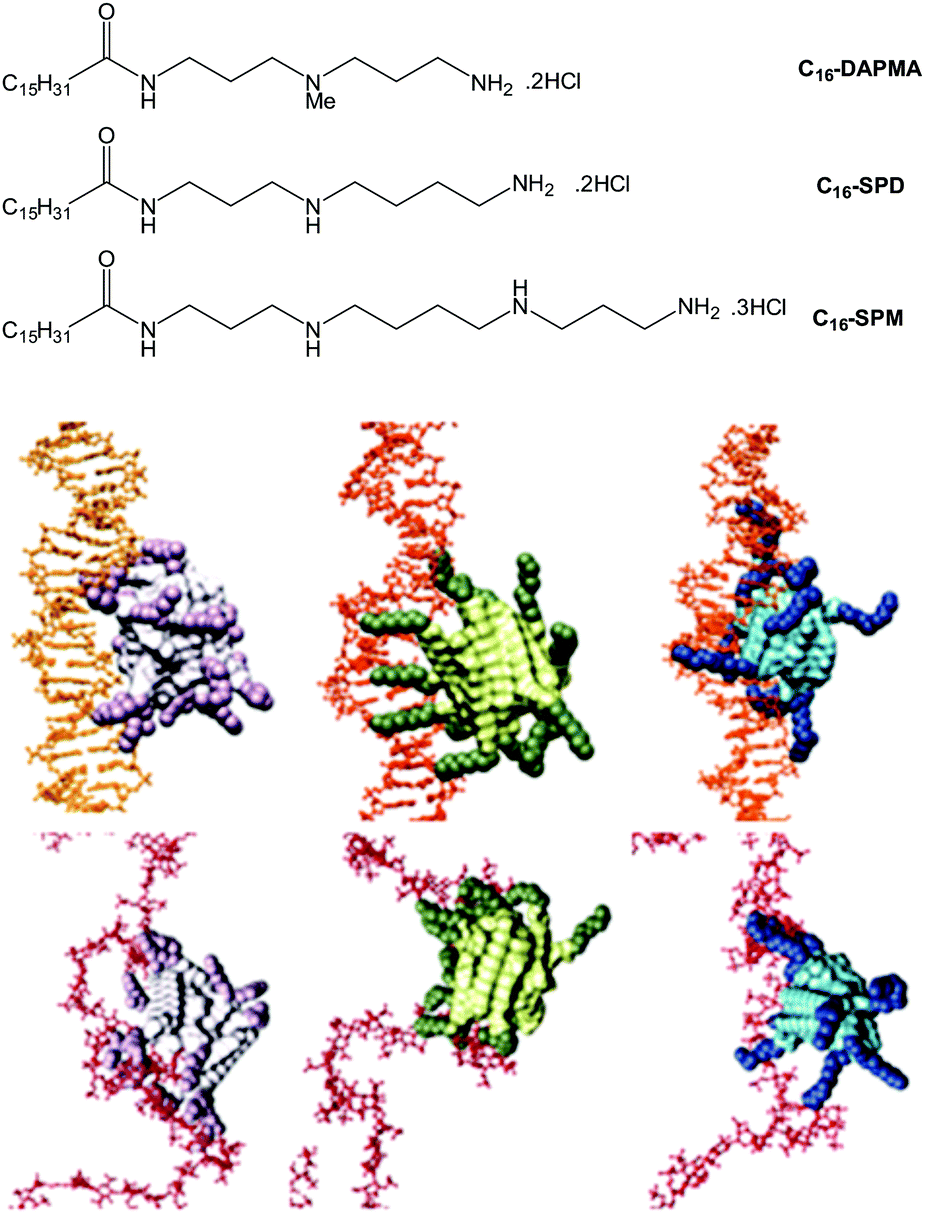 | ||
| Fig. 13 (top) Structures of compounds investigated as part of the ligand modification study, and (bottom) equilibrated atomistic molecular dynamics (MD) simulation snapshots of SAMul micelles binding DNA (upper panel, orange) and heparin (lower panel, firebrick), from left to right: C16-DAPMA (light grey (C16) and plum (DAPMA)), C16-SPD (lime green (C16) and forest green (SPD)), and C16-SPM (steel blue (C16) and navy blue (SPM)). Hydrogen atoms, water molecules, ions and counterions are not shown for clarity. The shape persistence of DNA which favours C16-SPM and more adaptive nature of heparin favouring C16-SPD are evident in this modelling study. | ||
The hydrophobic unit can also affect the SAMul binding event. To demonstrate this, we synthesised a family of molecules with the same cationic ligand (DAPMA) and a hydrophobic aliphatic group with eighteen carbon atoms, but having one, two, or three alkenes within the hydrophobic chain (C18-1, C18-2 and C18-3, Fig. 14).71 The presence of more alkenes led to geometric distortion, yielding larger self-assembled multivalent (SAMul) nanostructures and hence modifying the ligand display. Polyanion binding was studied using dye displacement assays and ITC, with data in agreement that heparin bound most effectively to C18-1, and DNA to C18-3, even though the molecular-scale structural differences of the SAMul systems are buried in the hydrophobic core. Multiscale modelling suggested that adaptive heparin somewhat folds around smaller C18-1 assemblies maximising enthalpically favourable interactions, while ‘shape-persistent’ DNA cannot easily bend, and forms a similar number of interactions with each assembly, but with less entropic cost for binding to the larger flatter surface of C18-3. Once again, this provides fundamental insight into electrostatic molecular recognition in biologically relevant systems and suggests not all polyanions are the same.
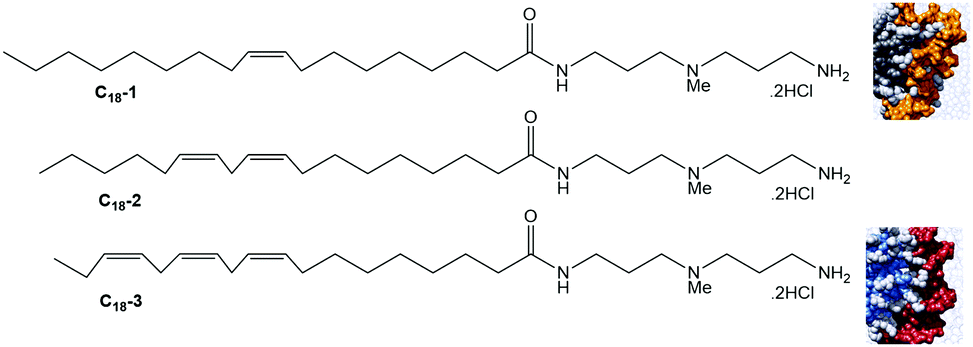 | ||
| Fig. 14 Structures of SAMul systems used to study the impact of the hydrophobic unit on ligand display and multivalent binding. C18-1 (grey) forms a more tightly packed assembly which adaptive heparin (orange) is better able to wrap around, while the larger assemblies formed by C18-3 (blue/grey) are more compatible with maximising interactions with shape persistent DNA (red) that cannot bend around the SAMul nanostructure. | ||
The impact of buffer on SAMul electrostatic recognition has also been studied.72 ITC and dye displacement assays indicated that the binding of C16-DAPMA to heparin was significantly stronger in Tris–HCl > HEPES > phosphate. This results from interactions between the anionic buffer component (phosphate/sulfonate/chloride) and the cationic binder. Such interactions occur in 10 mM buffer, even in the presence of 150 mM NaCl. Furthermore, even a ‘Good’ buffer such as HEPES competes at the binding interface. The impact of buffer on SAMul nanostructures was even more significant than on a simple heparin binder such as MalB. ITC was used to determine the difference in ΔG for heparin binding between Tris–HCl and phosphate buffer. This buffer induced difference was 1.20 kcal mol−1 for MalB (14.1% of the total) but rose to 1.77 kcal mol−1 for C16-DAPMA (21.9% of the total). The greater adverse effect of competitive buffers on SAMul systems arose from a decrease in the larger enthalpic term resulting from the highly charged multivalent binding interface. This study highlighted the importance of choosing even often-overlooked species, such as buffers,73 very carefully in molecular recognition studies and demonstrated that highly charged nanoscale binding interfaces are sensitive to buffer.
Once again, by starting with an application in mind, we have uncovered new fundamental concepts. This has led to the emergence of ideas such as shape-persistent and adaptive multivalency. Furthermore, the medical drive behind our work has meant all of our anion binding studies have transitioned from organic solvents into, as a minimum, buffered water with high levels of competitive salt. We believe that understanding binding interactions in highly competitive and complex media (e.g. buffers, human serum or blood), with multiple competing influences – will emerge as a key theme of supramolecular chemistry over the coming 10 years, with many more chemists starting to explore this fascinating but challenging ‘real world’.
From organogels to hydrogels – pursuing applications in drug delivery
Another thing that became apparent watching major transplant surgery, was the need for large amounts of pharmaceutical intervention, such as painkillers, anti-rejection drugs and a wide range of supplements. After surgery, such drugs are mostly delivered intravenously, and then later orally. Reflecting on the extensive drug treatment made me reflect on different modes of drug delivery, which may be more effective for long-term treatment or controlled drug release. In particular, my research team had been developing extensive expertise in working with self-assembled gel-phase materials74 which form as a result of non-covalent interactions assembling molecular-scale building blocks in a ‘1-dimensional’ manner into extended nanofibres, that ultimately form a sample spanning network which manifests itself macroscopically as a gel (Fig. 15).75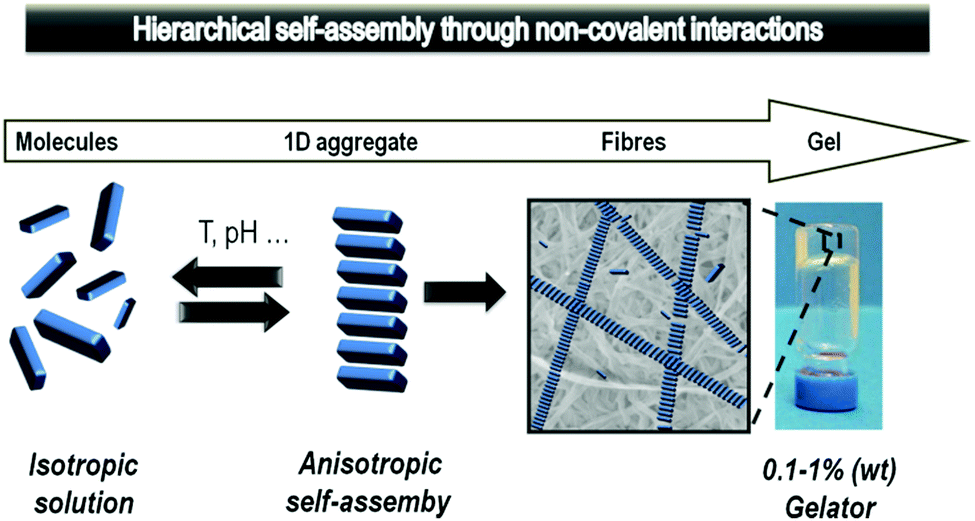 | ||
| Fig. 15 Schematic diagram of the self-assembly of gels from molecular-scale building blocks assembling into fibrils, which aggregate into fibres and ultimately for a sample-spanning network with macroscopic gel-phase behaviour. | ||
Our fundamental research on ‘supramolecular gels’ had, for example, demonstrated that such materials were highly responsive – capable of selecting and releasing specific components from complex mixtures76 and evolving their structures over time in response to different stimuli.77 We realised that these would be useful features in materials for drug delivery. There was just one problem – all of our long-established gels were only stable in organic solvents such as toluene, being held together by intermolecular hydrogen bond interactions between peptide building blocks. The presence of competitive media was simply too disruptive, leading to fibril disassembly and gel breakdown. We therefore set ourselves the challenge of developing a simple new commercially-viable hydrogelator that operated in water. Such a system could open up a range of biomedical (and other) applications.78
For inspiration, we turned to one of the best known families of gelator – 1,3:2,4-dibenzylidenesorbitol (DBS) – about which we wrote a key review.79 First reported by Meunier in 1891 as a product of the reaction between sorbitol and two equivalents of benzaldehyde,80 DBS gels have been widely applied in an industrial setting over the past century. DBS has been self-assembled in polymers to enhance transparency.81 It has also been used as the gelation component in personal care products such as deodorant sticks.82 However, DBS is always used in organic solvents or polymer melts. In pure water, it struggles to self-assemble into an effective gel owing to its poor solubility – indeed, solvent effects on DBS self-assembly have recently been studied in detail.83 DBS is often described as a butterfly-like surfactant, with the sorbitol unit as the body and the aromatic rings as the wings. It assembles into fibrils as a result of intermolecular hydrogen bond interactions via the body and solvophobic/π–π interactions between the wings (Fig. 16), with the balance depending on the solvent in which the gelator assembles.
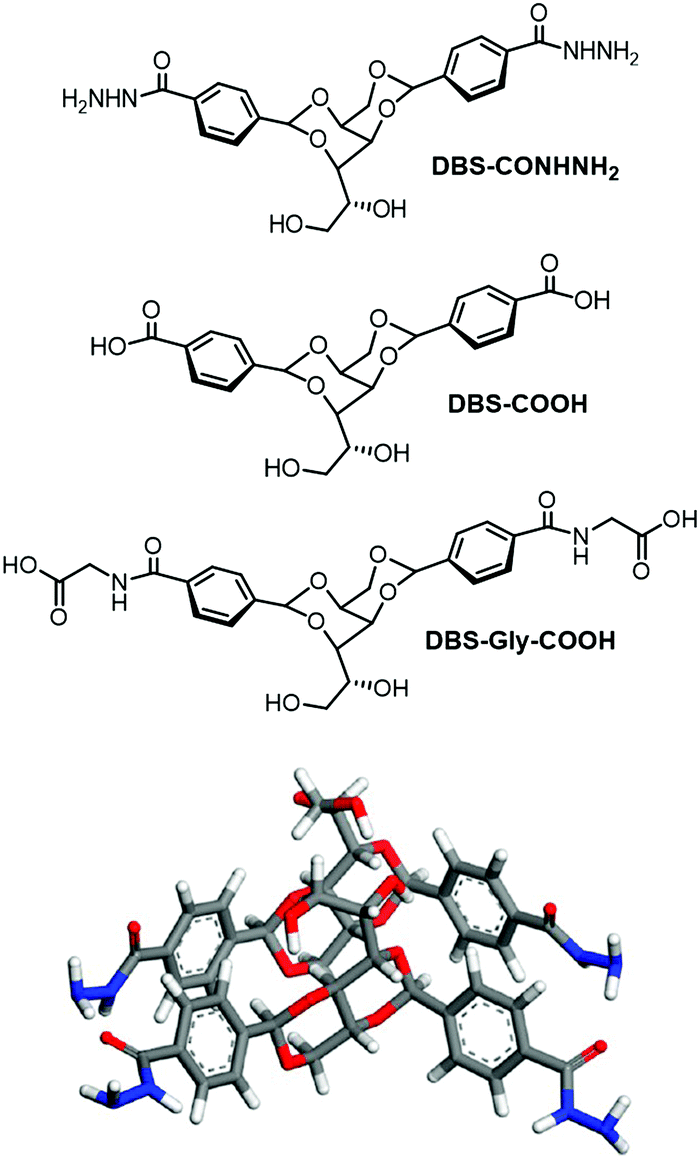 | ||
| Fig. 16 (top) New DBS hydrogelators DBS-COHNH2, DBS-COOH and DBS-Gly-COOH developed by us, and (bottom) self-assembly mode of DBS-CONHNH2 as modelled by Knani and Alperstein.88 | ||
We therefore decided to target hydrogels using this simple, yet versatile, molecular framework. Derivatives of DBS were reasonably well-known, as benzaldehyde can simply be replaced with a substituted benzaldehyde during synthesis. However, only simple substituents (e.g. Me, F, etc.) had been used to modify the performance for a specific industrial application.84 We wanted to insert functional groups that might increase the solubility of DBS in water, hence opening the possibility of hydrogel formation. Surprisingly, this had not previously been reported. Using a methyl ester of 4-carboxybenzaldehyde gave access to DBS-COOMe, and inserted a functional handle onto DBS that could be simply converted into an acid (DBS-COOH)85 or an acylhydrazide (DBS-CONHNH2)86 (Fig. 16). Pleasingly, this strategy worked and both of these compounds could be synthesised on large scale and could form hydrogels – a very significant step forwards for DBS chemistry. We further extended this family of hydrogels by modifying DBS-COOH to yield DBS-Gly-COOH.87 Following our discovery of these hydrogels, Knani and Alperstein's computer simulation suggested that hydrogen bonding between the sorbitol ‘bodies’ of the gelator were weakened by the presence of water, and that the aromatic rings were rigidified.88
The carboxylic acid, DBS-COOH, formed gels via protonation of the highly soluble carboxylate anion, yielding the less soluble carboxylic acid form, and allowing the ‘solid-like’ gel fibres to assemble with a degree of controlled kinetics in order to form a sample-spanning network.85 This was achieved using the well-established glucono-δ-lactone hydrolysis slow acidification method, as pioneered by the Adams group.89 The acylhydrazide, DBS-CONHNH2, was stable across the entire pH range from 2–12,86 and the solubility of this gelator was such that sample-spanning gels could be formed simply by heating and cooling a sample in water. Many well-established hydrogels are based on carboxylic acids, and require pH manipulation to form gels – we noted that acylhydrazides may potentially be effective carboxylic acid replacements in a wide-range of hydrogels.86 In this way, a new insight into gelators emerged from our application-driven hunt for new hydrogels.
We were then interested in the ability of these DBS-CONHNH2 gels to bind and release active agents with the ultimate goal of controlled drug release. The ability of this gel to extract pollutant dyes from model waste water was initially tested, as this is a quick and easy visual test – it was rapidly possible to screen the gel against a variety of different dyes in order to explore uptake selectivity.86 Dye extraction was controlled both by the structure of the dye, and pH. Fully-ionised dyes were less readily adsorbed onto the self-assembled gel fibres, presumably as a consequence of their higher water solubility. Changing pH altered the ionisation state of the dyes and hence provided a mechanism for desorbing dyes from the gel, meaning the gel could be used for repeat cycles of uptake and release. It is worth noting that this strategy is of great interest for remediating polluted water – indeed gels are highly effective in this application as a result of the nanoscale fibres having high relative surface areas, able to achieve very efficient uptake.90
Moving on from dyes towards biologically-relevant substances, we explored the ability of the DBS-CONHNH2 hydrogel to bind active pharmaceutical ingredients (APIs) – specifically, acid-functionalised non-steroidal anti-inflammatory drugs (NSAIDs), such as ibuprofen and naproxen.91 The gel could form in the presence of these APIs, and there were direct interactions between the acid on the API and the NH2 group displayed on the surface of the gel fibres – another form of self-assembled multivalent (SAMul) ligand display (Fig. 17). Interestingly, the APIs were immobilised at low pH values (<5), but once the pH was increased, they were desorbed from the gel, as the carboxylic acid became ionised and no longer interacted with the gel nanofibres. These hydrogels therefore achieved pH-triggered drug release of pharmaceutical relevance – the stomach has low pH (2–3) conditions, while the intestine, where drug release is most desirable is pH 7–8. As such, gels based on DBS-CONHNH2 can achieve targeted delivery of acid-functionalised APIs. Interestingly, naproxen causes well-documented side effects within the stomach, which can be particularly severe for patients who have longer-term use of such drugs, for example after major surgery.92 Most importantly, however, this study exemplified that self-assembled multivalent gel-phase materials can achieve controlled binding and release of a biologically active species in the presence of a triggering stimulus. We suggest that in the future, this might be of relevance to controlled delivery of a wide range of acid-functionalised APIs, such as statins.
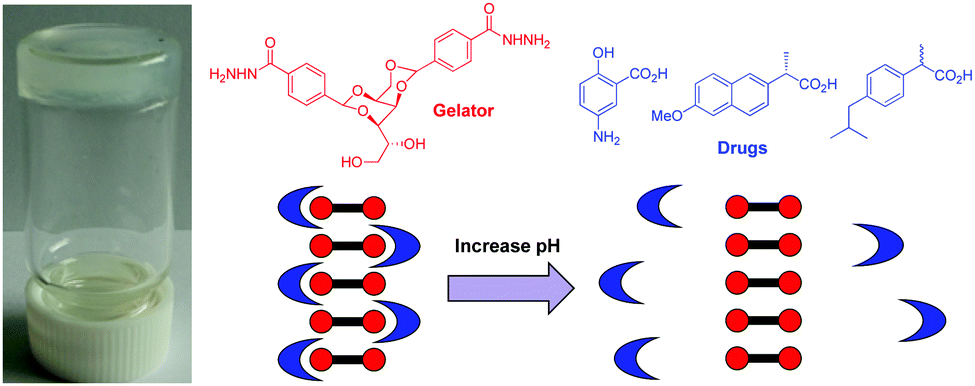 | ||
| Fig. 17 Image of hydrogel formed by DBS-CONHNH2 and naproxen. Structures of the gelator and some acid-functionalised non-steroidal anti-inflammatory drugs. Schematic of pH mediated drug uptake and release as a result of interactions with the self-assembled nanofibre surfaces. | ||
One disadvantage of self-assembled supramolecular hydrogels, such as DBS-CONHNH2, is that they are mechanically weak materials. This may be desirable for drug formulation for (e.g.) transdermal delivery, but is less suitable for delivery via an oral route. This weakness is a direct result of the reversible non-covalent forces that hold together self-assembled gels. In contrast, polymer gels can be significantly more robust owing to the covalent constitution of the network. As such, we reasoned that combining the two types of material into hybrid hydrogels would offer significant synergistic advantages (Fig. 18). To demonstrate this principle, we combined self-assembling low-molecular-weight gelator (LMWG) DBS-COOH with a typical polymer gelator (PG), agarose.85 The LMWG assembles on lowering pH, while the PG network forms by applying a heat–cool cycle. Using orthogonal methods of network formation is a useful tool for enabling their sequential formation. We demonstrated that the LMWG network could then be disassembled by raising pH, while the agarose PG network ensured the overall hybrid hydrogel remained intact. As such, these materials had both responsive and robust components. We reviewed this general approach to hybrid LMWG/PG gels, collecting together all the examples-to-date in a field-defining article.93
 | ||
| Fig. 18 Schematic of hybrid hydrogels formed by combination of a responsive self-assembling LMWG network and a more robust polymer gel (PG) network. | ||
Extending this approach, we recently combined DBS-CONHNH2 with a photo-initiated crosslinked polymer hydrogel based on poly(ethyleneglycol dimethacrylate) (PEGDM) and loaded the resulting gels with naproxen.94 The PEGDM provides mechanical strength and gives the hybrid hydrogel a self-supporting nature, while the self-assembled DBS-CONHNH2 network interacts with naproxen and controls its release depending on the pH of the surroundings. On exposing different sides of the gel to solutions with different pH values, directional release of the drug was achieved, controlled by the pH. We refer to this as directional release (Fig. 19). It opens the longer-term possibility of making gels with defined shapes in which the self-assembled network controls drug release in a directional manner under the influence of different stimuli towards a desired target. Such gels could, for example, be loaded with therapeutic agents and implanted into patients after surgery, releasing the drugs in a directional manner in response to biological stimuli. Further research in this direction is currently in progress in our labs.
 | ||
| Fig. 19 Directional release of naproxen from a hybrid hydrogel based on DBS-CONHNH2 and PEGDM with release of the drug occurring preferentially into a solution of pH 7 in comparison to one of pH 3. | ||
Smart multi-functional hydrogels as tissue engineering scaffolds to address transplantation problems
It is tempting to think that transplantation, as successfully achieved for Sam, is a solution to problems faced by patients with cystic fibrosis, but sadly this is not the case. Many CF patients die waiting for a transplant,95 and even for those patients lucky enough to receive a transplant, median life expectancy post-transplant remains stubbornly just 5–10 years – primarily a result of problems with chronic rejection.96 The only option for patients with this problem is a second transplant – but ethical and surgical considerations mean this is a rare event.97 Across all types of transplantation, the limiting factor is the lack of suitable donor organs. As such, regenerative medicine, and more specifically tissue engineering has emerged as a strategy for potentially growing organs and other tissue ex vivo and then implanting them into a patient.98 Given the ongoing revolution in stem cell technology, it is possible, at least in principle, to harvest stem cells from a patient, and then grow a replacement organ from these cells containing their own genetic material. Not only would this solve problems with organ supply, but it would eliminate the major problems associated with organ rejection and has the potential to transform lung transplant survival statistics.99 In order to grow an organ, a matrix/scaffold is required, and gels have emerged as one fascinating class of material that might be used.100 As soft wet materials, gels can be compatible with growing tissue and help direct cell growth – indeed the extracellular matrix is itself a type of gel.101 Given the potential transformative effects of such medicinal technology, the strong personal motivation given my husband's lung allograft rejection, and our existing expertise in self-assembled gels, we were impatient to get involved in this area of research.It is worth emphasising that much tissue engineering research has been carried out using polymer hydrogels.100 However, we (and others) reasoned that self-assembled low-molecular-weight gels may offer significant advantages for tissue engineering. Self-assembled materials have the advantage of being highly responsive, and they can disassemble into individual molecular-scale building blocks. This offers a significant advantage for 3D tissue engineering, in which tissue is grown within a gel, as the gel could be simply removed, and the tissue recovered.102 Some early work in this field achieved eye-catching in vivo results using self-assembled hydrogels based on relatively complex peptides, including nerve regeneration within the optic nerve and spinal cord, restoring function in disabled animal models.103 However, there has been less research making use of very simple low-molecular-weight gelators for tissue engineering.
In many ways, it is relatively straightforward to perform simple cell tissue culture on gels. What is more challenging, is to encourage the cells to do interesting and sophisticated things, which may be required for the growth of complex tissue such as organs. As such, our initial focus has been on adding functionality to our hydrogels so that not only can we grow tissue, but hopefully we can do it in smarter and more directed ways. In this way, heading towards our ultimate application, we reasoned that new fundamental insights would also emerge. In particular, we targeted incorporating the following features into LMWG hydrogels:
• Binding and release of active agents (this could help direct, control, or respond to, cell growth). Our work described in the previous section on controlled drug release is a step towards this goal.
• Conductive gels (tissue grown on such materials may then be stimulated electrically to achieve differential growth).104
• Spatially resolved multicomponent materials with multiple domains, each having different properties (this may facilitate spatially resolved and defined tissue growth).
These innovations would yield smarter LMWG hydrogels moving the state-of-the-art significantly forwards. In the long run, this may generate soft materials with the capacity to exert both temporal and spatial control over cell growth, directing biological outcomes, with the gels being capable of precise levels of intervention, and responsive to the growing tissue.
Controlled release
As described above, we have demonstrated that DBS-CONHNH2 can achieve controlled encapsulation and release of acid-functionalised drugs. We extended this work to develop gels capable of the encapsulation and release of heparin (Fig. 20).105 Heparin encourages the angiogenesis of growing tissue and also recruits other growth factors. As such, it can exert a significant influence on growing tissue.106 We combined LMWG DBS-COOH with PG agarose (to provide robustness) in a hybrid hydrogel formulation, along with heparin. The LMWG endows the hybrid hydrogel with pH-responsive behaviour, while the PG provides mechanical robustness. The rate of heparin release could be controlled through network density and composition, with the LMWG and PG behaving differently as a result of interactions between the LMWG and heparin. The addition of a micellar SAMul heparin binder (C16-DAPMA, described above) to the formulation completely inhibited heparin release through binding. Overall, this research demonstrated that a multi-component approach can yield exquisite control over self-assembled materials with all four components playing individual roles within the hybrid material. We reason that controlling orthogonality in such systems will underpin further development of controlled release systems for tissue engineering applications.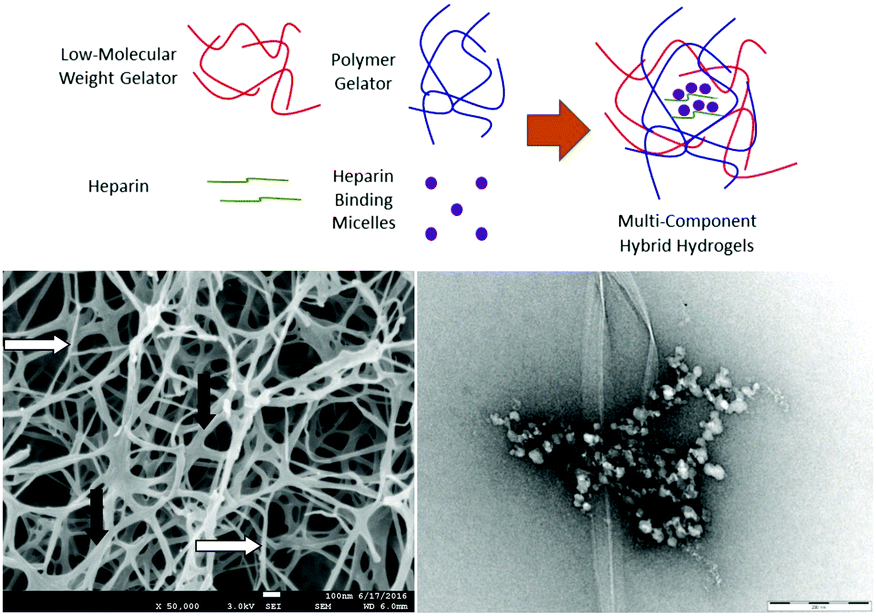 | ||
| Fig. 20 Hybrid hydrogel based on DBS-COOH (LMWG), agarose (PG), heparin and C16-DAPMA heparin-binding micelles. All four components could operate independently of one another. The SEM image of the hybrid hydrogel formed by DBS-COOH and agarose showed both larger fibres associated with DBS-COOH (black arrows) and smaller nanofibres associated with agarose (white arrows). The TEM image of the hybrid hydrogel in the presence of heparin and heparin-binding micelles showed both gel nanofibres and hierarchically-organised C16-DAPMA micelles bound to heparin. | ||
Conductive gels
With the target of conductive gels, we demonstrated that DBS-CONHNH2 selectively extracts precious metals from electronic waste.107 The acylhydrazides reduced these metals in close proximity to the gel nanofibres, and as a result, immobilised metal nanoparticles were generated, the vast majority of which were organised along the nanofibrillar network (Fig. 21). In the case of gold or silver, these gels had useful conductivities (Fig. 21). After partial drying to contract the network, the AuNP-loaded gel had a higher conductance than the equivalent hydrogel loaded with single walled carbon nanotubes. This gel was used to modify an electrode surface, which was functional for electrocatalysis. Given that hydrogels are often compatible with biological tissue, conductive gels offer a unique mechanism by which cells could be stimulated electrically.104 Work towards this challenging target is in progress.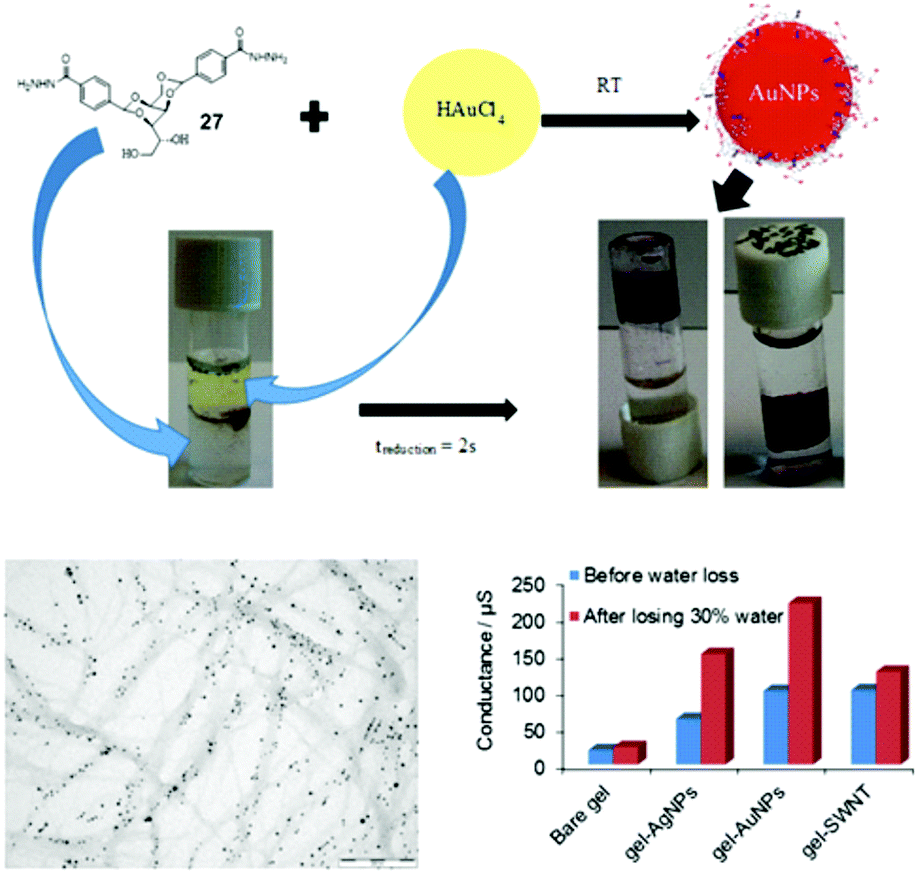 | ||
| Fig. 21 Use of DBS-CONHNH2 hydrogels to extract precious metals (e.g. Au) from mixtures with reduction to metal nanoparticles. TEM image demonstrates the organisation of metal nanoparticles on the nanofibre network. Graph demonstrates conductance of the DBS-CONHNH2 hydrogel formulated as a hybrid gel with agarose to give physical robustness, and hybrid hydrogels modified with AgNPs, AuNPs or SWNTs. On drying by 30% to bring AuNPs into close proximity, the AuNP-modified gels showed excellent conductances. | ||
Spatially resolved gels
Spatial resolution has considerable potential for tissue engineering, yet has been very rarely achieved with supramolecular gels,108 most of which are simply presented as gels formed in up-turned sample vials. In order to achieve spatial resolution within our gels, we turned for inspiration to the burgeoning field of polymer hydrogels – many of which are applied for tissue engineering.109 Although photo-activation is quite widely used in such materials as a way of achieving spatial control, perhaps surprisingly, it has been less wisely applied to self-assembled supramolecular gels to achieve spatial control. We initially developed a system in which we patterned a rigid crosslinked photo-initiated polymer hydrogel based on PEGDM within a soft DBS-COOH network using photo-patterning methods under a laser-jet-printed acetate mask, fabricating a multi-domain gel with hard regions (containing both PEGDM and DBS-COOH) and soft regions (containing only DBS-COOH) (Fig. 22).110 Dye diffusion was rapid through the soft supramolecular gel domain, but prevented by the rigid PEGDM network. This indicates that spatially resolved control over the diffusion and release of active ingredients should be possible. Indeed, the naproxen-loaded gel system described above (Fig. 19) was used to further demonstrate this principle.94 | ||
| Fig. 22 Spatially resolved hybrid hydrogel in which ‘Y-shaped’ PEGDM has been patterned into DBS-COOH. As can be seen, the DBS-COOH domain is very soft, while the PEGDM/DBS-COOH hybrid hydrogel is robust and can be lifted out of the gel. | ||
LMWG hydrogelator, DBS-Gly-COOH, opened up the possibility of patterning one self-assembled gel within another for the first time.87 The different pKa values of DBS-COOH (ca. 5.5) and DBS-Gly-COOH (ca. 4.5) mean they assemble sequentially as the pH is lowered. Careful control of conditions allowed us to form the DBS-COOH network using an equimolar chemical source of acid (glucono-δ-lactone), with the DBS-Gly-COOH network being subsequently assembled by activating a photo-acid (diphenyliodonium nitrate) under UV-stimulation. Performing the photo-acidification through a mask allowed us to ‘write’ a DBS-Gly-COOH network into a preformed DBS-COOH gel (Fig. 23). Interestingly, the first gel network is essential if the second network is to be patterned in with good spatial resolution. We reason that pattered multi-domain self-assembled LMWG gels such as these may spatially direct cell growth and then at a later stage, unlike many polymer gels, can be easily disassembled.
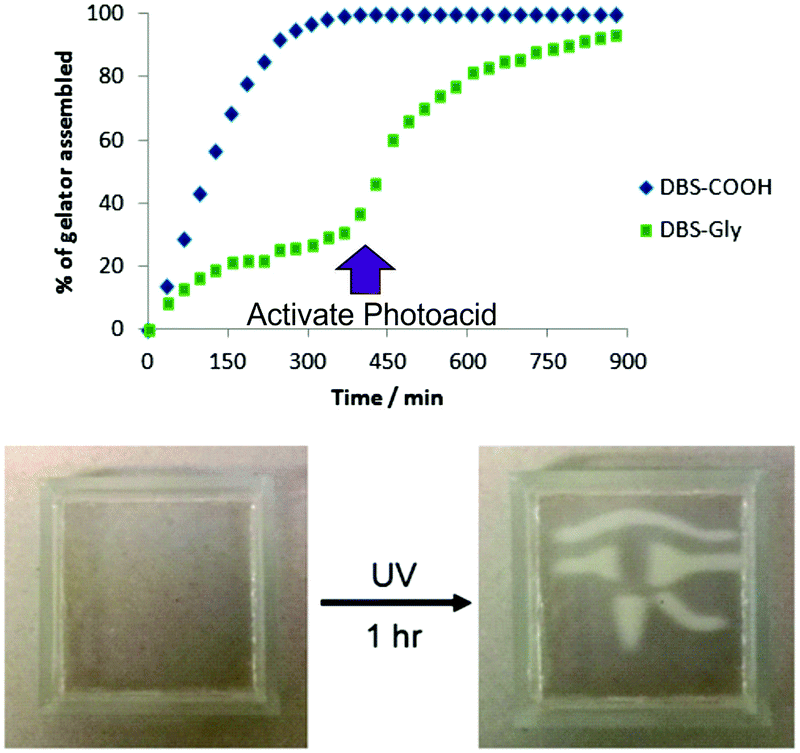 | ||
| Fig. 23 Graph reporting the results of NMR experiments demonstrating that DBS-COOH assembles in the presence of glucono-δ-lactone before DBS-Gly-COOH as a result of its higher pKa value. The DBS-Gly-COOH network then assembles second – which can be achieved by activation of the photoacid. Performing this latter process under a mask yields a material in which DBS-Gly-COOH is patterned into DBS-COOH with excellent spatial resolution. | ||
We suggest that combining controlled release, conductivity and spatial control within LMWGs yields a unique platform of technologies to develop innovative tissue engineering materials. To demonstrate these systems are compatible with tissue growth, experiments were performed with 3T3 Mouse Fibroblast cells, and it was shown that these could indeed grow effectively on simple DBS-CONHNH2.111 A major EPSRC grant is now enabling us to translate our fundamental gel technologies into controlled tissue engineering materials. Indeed, we look forward to continuing to develop new fundamental approaches and strategies in gel-phase materials and translating these into medicinal applications. We believe that explorations of smart self-assembled materials in biomedical applications will occupy much of our attention for the next 10 years and beyond.
Conclusions
In summary, this paper reflects on developments in my research group at York over the past 10 years in York. Inspired by the health problems of my husband, we have shifted some of our research into a more applied direction. All of our self-assembling systems are now being studied in aqueous, highly complex, media – a significant challenge for supramolecular chemistry. Furthermore, in the process of targeting new applications, we have developed enhanced theoretical understanding and initiated a number of new concepts in self-assembly science.• Flexible multivalency: screening and sacrifice. Flexibility in multivalent binding systems has advantages – e.g., flexible ligands can reorganise and shield electrostatic binding interfaces from competitive electrolyte.16
• Self-assembled multivalency (SAMul). Self-assembly can organise dynamic tunable multivalent ligand arrays.26,27,29a,54
• Temporary ‘on–off’ multivalency. Multivalent arrays that can degrade and/or disassemble enable high-affinity multivalency to be turned off, limiting persistence and potential toxicity.20,38,40
• Adaptive and shape persistent multivalency. Fundamental differences between the binding of polyanionic heparin and DNA suggest the former is adaptive and the latter shape-persistent.60,69,71
• Selective electrostatic binding. Electrostatic binding does not only depend on charge density – selectivity can be achieved based on the precise details of ligand structure.66,68,69
• Specific buffer effects. The choice of buffer, an often over-looked component, can impact on electrostatic binding, changing dissociation constants up-to 20-fold.72
• New DBS hydrogels. In the search for biorelevant systems, three new commercially-relevant hydrogelators based on 1,3:2,4-dibenzylidenesorbitol (DBS) have been developed.85–87
• Supramolecular controlled release. Gel nanofibres that form supramolecular interactions with (e.g.) drug molecules can control release profiles.91,94
• LMWG/PG hybrid hydrogels. Hybrid gels combining LMWG and PG gels can gain the advantage of synergistic effects from both networks.85,93,110
• Spatially resolved self-assembled hydrogels. Spatial resolution can fabricate complex self-assembled gels – moving LMWGs beyond being simple gels in vials.87,94,110
In addition to these fundamental insights and new strategic approaches, our research now approaches varied applications, and progress is ongoing in each of the following biomedically-relevant areas:
(i) Gene delivery. SAMul systems have been optimised in terms of morphology, and synergistic advantages of mixing different self-assembling units have been demonstrated. Optimised systems can achieve effective siRNA delivery in vitro and do not elicit an inflammatory response in vivo.
(ii) Coagulation control. Clotting times can be controlled in coagulation assays in human plasma using SAMul systems. Further stabilisation of self-assembled systems in highly competitive biological conditions is of key ongoing importance before translation in vivo.
(iii) Heparin sensing. Mallard Blue operates effectively in human serum/plasma and detects heparin using simple readouts. Further work to develop dual wavelength sensors is ongoing.
(iv) Drug formulation. Key drugs (e.g. naproxen) can be formulated in gels for controlled release. Gels are currently being tested in a range of settings, and for different modes of drug delivery, with directional release at controlled rates from shaped gels opening new paradigms in drug delivery.
(v) Tissue engineering. Combining release of active ingredients, conductivity and spatial resolution should encourage stem cells to grow in complex and sophisticated ways as a result of the chemistry programmed into self-assembled gels.
As illustrated by this article, the last ten years in our laboratory have been fruitful and stimulating. Approaching applied targets has not only provided a guiding principle behind some of our research, it has motivated young researchers, and inspired us to persevere through the inevitable string of paper and grant rejections. Furthermore, as well as heading towards these vital medical applications, we feel we have also become better fundamental scientists – developing new paradigms that are increasingly being applied in other scientists’ labs.
As scientists, we rarely talk about the personal – after all, the underpinning philosophy of science is that whoever performs the studies, the results will be the same. However, I strongly believe that personal experiences and interests can direct project development and enable intellectual connections between diverse areas of science in unique ways. The personal really does matter, and we need to support diverse scientists, so that diverse solutions to problems can be found.112 As I write this, given Sam's health problems, it is difficult to know what the future will bring personally, but I know that professionally, the decision to let the ‘personal’ influence my direction of academic travel is one that I will never regret.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
I would like to thank a research team that has included some spectacular talents – their names appear in the references, many have gone on to great things. The story told in this Feature Article does not, and cannot, do justice to all of the great work they have done, focussing as it does on the more applied aspects of our research. I would also like to thank marvellous collaborators from across the world who never fail to make our research better – Sabrina Pricl, Rainer Haag, Marcelo Calderon, Mauri Kostiainen, Bea Escuder, Julián Rodríguez-López, Ian Hamley, Dan Pack, Hak-Fun Chow and Jerry Turnbull have all contributed to the work discussed here. The research presented in this article has been supported by EPSRC (EP/PO3361X/1 and DTA awards), BBSRC (DTA awards), European Commission (316656 and 628757), The Wild Fund and The University of York. Finally, I must also thank Sam, for keeping me grounded and making me a better person in spite of everything he has been through – a personal journey I will never regret. This feature article is dedicated to him.Notes and references
- D. K. Smith, Chem. Commun., 2006, 34–44 RSC.
- J. S. Elborn, Lancet, 2016, 388, 2519–2531 CrossRef CAS.
- (a) A. R. Hirst, D. K. Smith, M. C. Feiters and H. P. M. Geurts, J. Am. Chem. Soc., 2003, 125, 9010–9011 CrossRef CAS PubMed; (b) A. R. Hirst, D. K. Smith, M. C. Feiters and H. P. M. Geurts, Chem. – Eur. J., 2004, 10, 5901–5910 CrossRef CAS PubMed; (c) B. Huang, A. R. Hirst, D. K. Smith, V. Castelletto and I. W. Hamley, J. Am. Chem. Soc., 2005, 127, 7130–7139 CrossRef CAS PubMed; (d) A. R. Hirst, D. K. Smith and J. P. Harrington, Chem. – Eur. J., 2005, 11, 6552–6559 CrossRef CAS PubMed.
- (a) D. K. Smith, Org. Biomol. Chem., 2003, 1, 3874–3877 RSC; (b) K. J. Winstanley and D. K. Smith, J. Org. Chem., 2007, 72, 2803–2815 CrossRef CAS PubMed.
- (a) D. K. Smith, J. Chem. Educ., 2005, 82, 393–400 CrossRef CAS; (b) D. A. Uhlenheuer, K. Petkau and L. Brunsveld, Chem. Soc. Rev., 2010, 39, 2817–2826 RSC; (c) P. A. Gale, J. T. Davis and R. Quesada, Chem. Soc. Rev., 2017, 46, 2497–2517 RSC.
- (a) L. Naldini, Nature, 2015, 526, 351–360 CrossRef CAS PubMed; (b) A. M. Keeler, M. K. ElMallah and T. R. Flotte, Clin. Transl. Sci., 2017, 10, 242–248 CrossRef CAS PubMed.
- See for example: (a) P. D. Beer, M. G. B. Drew, A. R. Graydon, D. K. Smith and S. E. Stokes, J. Chem. Soc., Dalton Trans., 1995, 403–408 RSC; (b) P. D. Beer, A. R. Graydon, A. O. M. Johnson and D. K. Smith, Inorg. Chem., 1997, 36, 2112–2118 CrossRef CAS PubMed; (c) P. D. Beer, J. Cadman, J. M. Lloris, R. Mártinez-Máñez, M. E. Padilla, T. Pardo, D. K. Smith and J. Soto, J. Chem. Soc., Dalton Trans., 1999, 127–133 RSC.
- (a) D. K. Smith and F. Diederich, Chem. Commun., 1998, 2501 RSC; (b) S. Koenig, L. Müller and D. K. Smith, Chem. – Eur. J., 2001, 7, 979–986 CrossRef CAS PubMed; (c) D. L. Stone, D. K. Smith and P. T. McGrail, J. Am. Chem. Soc., 2002, 124, 856–864 CrossRef CAS PubMed.
- (a) J. Haensler and F. C. Szoka, Bioconjugate Chem., 1993, 4, 372–379 CrossRef CAS; (b) J.-F. Kukowska-Latallo, A. U. Bielinska, J. Johnson, R. Spindler, D. A. Tomalia and J. R. Baker, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 4897–4902 CrossRef CAS PubMed; (c) J. S. Choi, E. J. Lee, Y. H. Choi, Y. J. Jeong and J. S. Park, Bioconjugate Chem., 1999, 10, 62–65 CrossRef CAS PubMed.
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E. W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS PubMed.
- (a) C. W. Tabor and H. Tabor, Annu. Rev. Biochem., 1984, 740–790 Search PubMed; (b) V. Vijayanathan, T. Thomas, A. Shirahata and T. J. Thomas, Biochemistry, 2001, 40, 13644–13651 CrossRef CAS PubMed.
- D. A. Lewenhoeck, Philos. Trans. R. Soc., 1677, 12, 1040–1046 CrossRef.
- (a) N. Korolev, A. P. Lyubartsev, L. Nordenskiöld and A. Laaksonen, J. Mol. Biol., 2001, 308, 907–917 CrossRef CAS PubMed; (b) Y. Burak, G. Ariel and D. Andelman, Curr. Opin. Colloid Interface Sci., 2004, 9, 53–58 CrossRef CAS.
- M. A. Kostiainen, J. G. Hardy and D. K. Smith, Angew. Chem., Int. Ed., 2005, 44, 2556–2559 CrossRef CAS PubMed.
- The following book collects a number of the outputs of this COST network: Dendrimers in Biomedical Applications, ed. B. Klajnert, L. Peng and V. Cena, RSC Publishing, Cambridge, 2013 Search PubMed.
- G. M. Pavan, A. Danani, S. Pricl and D. K. Smith, J. Am. Chem. Soc., 2009, 131, 9686–9694 CrossRef CAS PubMed.
- (a) G. R. Newkome and X. Lin, Macromolecules, 1991, 24, 1443–1444 CrossRef CAS; (b) G. R. Newkome, X. Lin and C. D. Weis, Tetrahedron: Asymmetry, 1991, 2, 957–960 CrossRef CAS; (c) C. M. Cordona, R. E. Gawley and C. Gawley, J. Org. Chem., 2002, 67, 1411–1413 CrossRef.
- (a) H. Ihre, A. Hult and E. Soederlind, J. Am. Chem. Soc., 1996, 118, 6388–6395 CrossRef CAS; (b) H. Ihre, A. Hult, J. M. J. Fréchet and I. Gitsov, Macromolecules, 1998, 31, 4061–4068 CrossRef CAS; (c) H. Ihre, O. L. Padilla de Jesus and J. M. J. Fréchet, J. Am. Chem. Soc., 2001, 123, 5908 CrossRef CAS PubMed.
- (a) E. R. Gillies and J. M. J. Fréchet, J. Am. Chem. Soc., 2002, 124, 14137–14146 CrossRef CAS PubMed; (b) E. R. Gillies, E. Dy, J. M. J. Fréchet and F. C. Szoka, Mol. Pharmaceutics, 2005, 2, 129–138 CrossRef CAS PubMed; (c) C. C. Lee, E. R. Gillies, M. E. Fox, S. J. Guillaudeu, J. M. J. Fréchet, E. E. Dy and F. C. Szoka, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16649–16654 CrossRef CAS PubMed.
- D. J. Welsh, S. P. Jones and D. K. Smith, Angew. Chem., Int. Ed., 2009, 48, 4047–4051 CrossRef CAS PubMed.
- (a) R. Duncan and L. Izzo, Adv. Drug Delivery Rev., 2005, 57, 2215–2237 CrossRef CAS PubMed; (b) H. Lv, S. Zhang, B. Wang, S. Cui and J. Yan, J. Controlled Release, 2006, 114, 100–109 CrossRef CAS PubMed; (c) J.-h. S. Kuo and Y. L. Lin, J. Biotechnol., 2007, 129, 383–390 CrossRef CAS PubMed; (d) J.-h. S. Kuo, M.-j. Liou and H.-c. Chiu, Mol. Pharmaceutics, 2010, 7, 805–814 CrossRef CAS PubMed.
- M. A. Kostiainen, G. R. Szilvay, D. K. Smith, M. B. Linder and O. Ikkala, Angew. Chem., Int. Ed., 2006, 45, 3538–3542 CrossRef CAS PubMed.
- S. P. Jones, G. M. Pavan, A. Danani, S. Pricl and D. K. Smith, Chem. – Eur. J., 2010, 16, 4519–4532 CrossRef CAS PubMed.
- P. Zhang and E. Wagner, Top. Curr. Chem., 2017, 375, 26 CrossRef PubMed.
- (a) R. Srinivas, S. Samanta and A. Chaudhuri, Chem. Soc. Rev., 2009, 38, 3326–3338 RSC; (b) S. Rietwyk and D. Peer, ACS Nano, 2017, 11, 7572–7586 CrossRef CAS PubMed.
- S. P. Jones, N. P. Gabrielson, D. W. Pack and D. K. Smith, Chem. Commun., 2008, 4700–4702 RSC.
- P. Posocco, S. Pricl, S. Jones, A. Barnard and D. K. Smith, Chem. Sci., 2010, 1, 393–404 RSC.
- J. E. Kingerywood, K. W. Williams, G. B. Sigal and G. M. Whitesides, J. Am. Chem. Soc., 1992, 114, 7303–7305 CrossRef CAS.
- (a) A. Barnard and D. K. Smith, Angew. Chem., Int. Ed., 2012, 51, 6572–6581 CrossRef CAS PubMed . Another key review in this area shortly followed our own: ; (b) K. Petkau-Milroy and L. Brunsveld, Org. Biomol. Chem., 2013, 11, 219–232 RSC.
- S. P. Jones, N. P. Gabrielson, C.-H. Wong, H.-F. Chow, D. W. Pack, P. Posocco, M. Fermeglia, S. Pricl and D. K. Smith, Mol. Pharmaceutics, 2011, 8, 416–429 CrossRef CAS PubMed.
- J. G. Hardy, M. A. Kostiainen, D. K. Smith, N. P. Gabrielson and D. W. Pack, Bioconjugate Chem., 2006, 17, 172–178 CrossRef CAS PubMed.
- O. Zelphati and F. C. Szoka, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 11493–11498 CrossRef CAS.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- I. S. Zuhorn, J. B. F. N. Engberts and D. Hoekstra, Eur. Biophys. J. Biophys. Lett., 2007, 36, 349–362 CrossRef CAS PubMed.
- R. P. Harbottle, R. G. Cooper, S. L. Hart, A. Ladhoff, T. McKay, A. M. Knight, E. Wagner, A. D. Miller and C. Coutelle, Hum. Gene Ther., 1998, 9, 1037–1047 CrossRef CAS PubMed.
- D. J. Welsh and D. K. Smith, Org. Biomol. Chem., 2011, 9, 4795–4801 CAS.
- D. J. Welsh, P. Posocco, S. Pricl and D. K. Smith, Org. Biomol. Chem., 2013, 11, 3177–3186 CAS.
- A. Barnard, P. Posocco, S. Pricl, M. Calderon, R. Haag, M. E. Hwang, V. W. T. Shum, D. W. Pack and D. K. Smith, J. Am. Chem. Soc., 2011, 133, 20288–20300 CrossRef CAS PubMed.
- A. Barnard, M. Calderon, A. Tschiche, R. Haag and D. K. Smith, Org. Biomol. Chem., 2012, 10, 8403–8409 CAS.
- A. Barnard, P. Posocco, M. Fermeglia, A. Tschiche, M. Calderon, S. Pricl and D. K. Smith, Org. Biomol. Chem., 2014, 12, 446–455 CAS.
- W. Fischer, M. A. Quadir, A. Barnard, D. K. Smith and R. Haag, Macromol. Biosci., 2011, 11, 1736–1746 CrossRef CAS PubMed.
- A. Tschiche, A. M. Staedtler, S. Malhotra, H. Bauer, C. Böttcher, S. Sharbati, M. Calderón, M. Koch, T. M. Zollner, A. Barnard, D. K. Smith., R. Einspanier, N. Schmidt and R. Haag, J. Mater. Chem. B, 2014, 2, 2153–2167 RSC.
- (a) N. Bessis, F. J. Garcia Cozar and M.-C. Boissier, Gene Ther., 2004, 11, S10–S17 CrossRef CAS PubMed; (b) X. Zhao, X. Li, Y. Zhao, Y. Cheng, Y. Yang, Z. Fang, Y. Xie, Y. Liu, Y. Chen, Y. Ouyang and W. Yuan, Front. Pharmacol., 2017, 8, 510 CrossRef PubMed.
- (a) X. Liu, J. Zhou, T. Yu, C. Chen, Q. Cheng, K. Sengupta, Y. Huang, H. Li, C. Liu, Y. Wang, P. Posocco, M. Wang, Q. Cui, S. Giorgio, M. Fermeglia, F. Qu, S. Pricl, Y. Shi, Z. Liang, P. Rocchi, J. J. Rossi and L. Peng, Angew. Chem., Int. Ed., 2014, 53, 11822–11827 CrossRef CAS PubMed; (b) C. Chen, P. Posocco, X. Liu, Q. Cheng, E. Laurini, J. Zhou, C. Liu, Y. Wang, J. Tang, V. Dal Col, T. Yu, S. Giorgio, M. Fermeglia, F. Qu, Z. Liang, J. J. Rossi, M. Liu, P. Rocchi, S. Pricl and L. Peng, Small, 2016, 12, 3667–3676 CrossRef CAS PubMed.
- (a) R. Barbucci, A. Magnani, S. Lamponi and A. Albanese, Polym. Adv. Technol., 1996, 7, 675–685 CrossRef CAS; (b) D. L. Rabenstein, Nat. Prod. Rep., 2002, 19, 312–331 RSC; (c) S. Middeldorp, Thromb. Res., 2008, 122, 753–762 CrossRef CAS PubMed.
- R. Balhorn, Genome Biol., 2007, 8, 227 CrossRef PubMed.
- (a) M. Nybo and J. S. Madsen, Basic Clin. Pharmacol. Toxicol., 2008, 103, 192–196 CrossRef CAS PubMed; (b) Y.-Q. Chu, L.-J. Cai, D.-C. Jiang, D. Jia, S.-Y. Yan and Y.-Q. Wang, Clin. Ther., 2010, 32, 1729–1732 CrossRef PubMed.
- C. Hermans and D. Claeys, Curr. Med. Res. Opin., 2006, 22, 471–481 CrossRef CAS PubMed.
- Selected examples: (a) M. Kikura, M. K. Lee and J. H. Levy, Anesth. Analg., 1996, 83, 223–227 CrossRef CAS PubMed; (b) S. Choi, D. J. Clements, V. Pophristic, I. Ivanov, S. Vemparala, J. S. Bennett, M. L. Klein, J. D. Winkler and W. F. DeGrado, Angew. Chem., Int. Ed., 2005, 44, 6685–6689 CrossRef CAS PubMed; (c) T. Mecca, G. M. L. Consoli, C. Geraci, R. La Spina and F. Cunsolo, Org. Biomol. Chem., 2006, 4, 3763–3768 RSC; (d) K. Rajangam, H. A. Behanna, M. J. Hui, X. Han, J. F. Hulvat, J. W. Lomansey and S. I. Stupp, Nano Lett., 2006, 6, 2086–2090 CrossRef CAS PubMed; (e) G. L. Montalvo, Y. Zhang, T. M. Young, M. J. Costanzo, K. B. Freeman, J. Wang, D. J. Clements, E. Magavern, R. W. Kavash, R. W. Scott, D. H. Liu and W. F. DeGrado, ACS Chem. Biol., 2014, 9, 967–975 CrossRef CAS PubMed; (f) S. Valimaki, A. Khakalo, A. Ora, L.-S. Johansson, O. J. Rojas and M. A. Kostiainen, Biomacromolecules, 2016, 17, 2891–2900 CrossRef CAS PubMed.
- Selected examples: (a) Z. L. Zhong and E. V. Anslyn, J. Am. Chem. Soc., 2002, 124, 9014–9015 CrossRef CAS PubMed; (b) A. T. Wright, Z. Zhong and E. V. Anslyn, Angew. Chem., Int. Ed., 2005, 44, 5679–5682 CrossRef CAS PubMed; (c) W. Sun, H. Bandmann and T. Schrader, Chem. – Eur. J., 2007, 13, 7701–7707 CrossRef CAS PubMed; (d) T. Briza, Z. Kejik, I. Cisarova, J. Kralova, P. Martasek and V. Kral, Chem. Commun., 2008, 1901–1903 RSC; (e) S. Wang and Y.-T. Chang, Chem. Commun., 2008, 1173–1175 RSC; (f) M. Wang, D. Q. Zhang, G. X. Zhang and D. B. Zhu, Chem. Commun., 2008, 4469–4471 RSC; (g) L.-J. Chen, Y.-Y. Ren, N.-W. Wu, B. Sun, J.-Q. Ma, L. Zhang, H. Tan, M. Liu, X. Li and H.-B. Yang, J. Am. Chem. Soc., 2015, 137, 11725–11735 CrossRef CAS PubMed; (h) M. Yang, J. Chen, H. Zhou, W. Li, Y. Wang, J. Li, C. Zhang, C. Zhou and C. Yu, Biosens. Bioelectron., 2016, 75, 404–410 CrossRef CAS PubMed.
- S. M. Bromfield, E. Wilde and D. K. Smith, Chem. Soc. Rev., 2013, 42, 9184–9195 RSC.
- A. C. Rodrigo, A. Barnard, J. Cooper and D. K. Smith, Angew. Chem., Int. Ed., 2011, 50, 4675–4679 CrossRef CAS PubMed.
- V. M. P. Vieira, V. Liljeström, P. Posocco, E. Laurini, S. Pricl, M. A. Kostiainen and D. K. Smith, J. Mater. Chem. B, 2017, 5, 341–347 RSC.
- S. M. Bromfield, P. Posocco, C. W. Chan, M. Calderon, S. E. Guimond, J. E. Turnbull, S. Pricl and D. K. Smith, Chem. Sci., 2014, 5, 1484–1492 RSC.
- (a) S. Tenjarla, Crit. Rev. Ther. Drug Carrier Syst., 1999, 16, 461–521 CrossRef CAS PubMed; (b) S. Gupta and S. P. Moulik, J. Pharm. Sci., 2008, 97, 22–45 CrossRef CAS PubMed; (c) S. Kim, Y. Shi, J. Y. Kim, K. Park and J.-X. Cheng, Expert Opin. Drug Delivery, 2010, 7, 49–56 CrossRef CAS PubMed; (d) M. J. Lawrence and G. D. Rees, Adv. Drug Delivery Rev., 2012, 64, 175–193 CrossRef.
- A. C. Rodrigo, S. M. Bromfield, E. Laurini, P. Posocco, S. Pricl and D. K. Smith, Chem. Commun., 2017, 53, 6335–6338 RSC.
- (a) M. D. Klein, R. A. Drongowski, R. J. Linhardt and R. S. Langer, Anal. Biochem., 1982, 124, 59–64 CrossRef CAS PubMed; (b) Q. Jiao and Q. Liu, Anal. Lett., 1998, 31, 1311–1323 CrossRef CAS.
- S. M. Bromfield, A. Barnard, P. Posocco, M. Fermeglia, S. Pricl and D. K. Smith, J. Am. Chem. Soc., 2013, 135, 2911–2914 CrossRef CAS PubMed.
- S. M. Bromfield, P. Posocco, M. Fermeglia, S. Pricl, J. Rodríguez-López and D. K. Smith, Chem. Commun., 2013, 49, 4830–4832 RSC.
- S. M. Bromfield, P. Posocco, M. Fermeglia, J. Tolosa, A. Herreros-López, S. Pricl, J. Rodríguez-López and D. K. Smith, Chem. – Eur. J., 2014, 20, 9666–9674 CrossRef CAS PubMed.
- C. W. Chan and D. K. Smith, Chem. Commun., 2016, 52, 3785–3788 RSC.
- (a) H. Szelke, S. Schübel, J. Harenberg and R. Krämer, Bioorg. Med. Chem. Lett., 2010, 20, 1445–1447 CrossRef CAS PubMed; (b) D. Li and N. J. Wagner, J. Am. Chem. Soc., 2013, 135, 17547–17555 CrossRef CAS PubMed.
- For a recent example see: S. Mishra and A. R. Horswill, mSphere, 2017, 2, e00135 CrossRef CAS PubMed.
- L. S. Jones, B. Yazzie and C. R. Middaugh, Mol. Cell. Proteomics, 2004, 3, 746–769 CAS.
- (a) M. Torrent, M. Victòria Nogués, D. Andreu and E. Boix, PLoS One, 2012, 7, e42692 CAS; (b) P. D. Mosier, C. Krishnasamy, G. E. Kellogg and U. R. Desai, PLoS One, 2012, 7, e48632 CAS.
- S. M. Bromfield and D. K. Smith, J. Am. Chem. Soc., 2015, 137, 10056–10059 CrossRef CAS PubMed.
- Selected examples: (a) X. G. Qu, J. O. Trent, I. Fokt, W. Priebe and J. B. Chaires, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 12032–12037 CrossRef CAS PubMed; (b) H. Nishi and Y. Kuwahara, J. Biochem. Biophys. Methods, 2001, 48, 89–102 CrossRef CAS PubMed.
- C. W. Chan, E. Laurini, P. Posocco, S. Pricl and D. K. Smith, Chem. Commun., 2016, 52, 10540–10543 RSC.
- L. E. Fechner, B. Albanyan, V. M. P. Vieira, E. Laurini, P. Posocco, S. Pricl and D. K. Smith, Chem. Sci., 2016, 7, 4653–4659 RSC.
- M. C. Z. Meneghetti, A. J. Hughes, T. R. Rudd, H. B. Nader, A. K. Powell, E. A. Yates and M. A. Lima, J. R. Soc., Interface, 2015, 12, 20150589 CrossRef PubMed.
- B. Albanyan, E. Laurini, P. Posocco, S. Pricl and D. K. Smith, Chem. – Eur. J., 2017, 23, 6391–6397 CrossRef CAS PubMed.
- A. C. Rodrigo, E. Laurini, V. M. P. Vieira, S. Pricl and D. K. Smith, Chem. Commun., 2017, 53, 11580–11583 RSC.
- C. M. H. Ferreira, I. S. S. Pinto, E. V. Soares and H. M. V. M. Soares, RSC Adv., 2015, 5, 30989–31003 RSC.
- A. R. Hirst, I. A. Coates, T. R. Boucheteau, J. F. Miravet, B. Escuder, V. Castelletto, I. W. Hamley and D. K. Smith, J. Am. Chem. Soc., 2008, 130, 9113–9121 CrossRef CAS PubMed.
- (a) R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed; (b) E. R. Draper and D. J. Adams, Chem, 2017, 3, 390–410 CrossRef CAS.
- A. R. Hirst, J. F. Miravet, B. Escuder, L. Noirez, V. Castelletto, I. W. Hamley and D. K. Smith, Chem. – Eur. J., 2009, 15, 372–379 CrossRef CAS PubMed.
- (a) W. Edwards and D. K. Smith, J. Am. Chem. Soc., 2013, 135, 5911–5920 CrossRef CAS PubMed; (b) W. Edwards and D. K. Smith, J. Am. Chem. Soc., 2014, 136, 1116–1124 CrossRef CAS PubMed.
- K. J. Skilling, F. Citossi, T. D. Bradshaw, M. Ashford, B. Kellam and M. Marlow, Soft Matter, 2014, 10, 237–256 RSC.
- B. O. Okesola, V. M. P. Vieira, D. J. Cornwell, N. K. Whitelaw and D. K. Smith, Soft Matter, 2015, 11, 4768–4787 RSC.
- M. J. Meunier, Ann. Chim. Phys., 1891, 22, 412 Search PubMed.
- (a) K. Hamada and H. Uchiyama, US Pat., 4016118, 1977 Search PubMed; (b) R. L. Mahaffey, US Pat., 4371645, 1983 Search PubMed; (c) H. Uchiyama, US Pat., 4483952, 1984 Search PubMed.
- (a) E. L. Roehl and H. B. Tan, US Pat., 4154816, 1979 Search PubMed; (b) E. L. Roehl, US Pat., 4346079, 1982 Search PubMed; (c) T. Schamper, M. Jablon, M. H. Randhawa, A. Senatore and J. D. Warren, J. Soc. Cosmet. Chem., 1986, 37, 225–231 CAS; (d) A. Esposito, T. Schamper and E. C. Henry, US Pat., 7799318, 2010 Search PubMed; (e) G. Malle and T. Luukas, US Pat., 9604992, 2010 Search PubMed.
- K. K. Diehn, H. Oh, R. Hashemipour, R. G. Weiss and S. R. Raghavan, Soft Matter, 2014, 10, 2632–2640 RSC.
- Z. Horváth, B. Gyarmati, A. Manyhárd, P. Doshev, M. Gahleitner, J. Varga and B. Puzánsky, RSC Adv., 2014, 4, 19737–19745 RSC.
- D. J. Cornwell, B. O. Okesola and D. K. Smith, Soft Matter, 2013, 9, 8730–8736 RSC.
- B. O. Okesola and D. K. Smith, Chem. Commun., 2013, 49, 11164–11166 RSC.
- D. J. Cornwell, O. J. Daubney and D. K. Smith, J. Am. Chem. Soc., 2015, 137, 15486–15492 CrossRef CAS PubMed.
- D. Knani and D. Alperstein, J. Phys. Chem. A, 2017, 121, 1113–1120 CrossRef CAS PubMed.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856–1862 RSC.
- B. O. Okesola and D. K. Smith, Chem. Soc. Rev., 2016, 45, 4226–4251 RSC.
- E. J. Howe, B. O. Okesola and D. K. Smith, Chem. Commun., 2015, 51, 7451–7454 RSC.
- N. Bhala, J. Emberson, A. Merhi, S. Abramson, N. Arber, J. A. Baron, C. Bombardier, C. Cannon, M. E. Farkouh, G. A. FitzGerald, P. Goss, H. Halls, E. Hawk, C. Hawkey, C. Hennekens, M. Hochberg, L. E. Holland, P. M. Kearney, L. Laine, A. Lanas, P. Lance, A. Laupacis, J. Oates, C. Patrono, T. J. Schnitzer, S. Solomon, P. Tugwell, K. Wilson, J. Wittes and C. Baigent, Lancet, 2013, 382, 769–779 CrossRef CAS.
- D. J. Cornwell and D. K. Smith, Mater. Horiz., 2015, 2, 279–293 RSC.
- P. R. A. Chivers and D. K. Smith, Chem. Sci., 2017, 8, 7218–7227 RSC.
- R. A. Belkin, N. R. Henig, L. G. Singer, C. Chaparro, R. C. Rubenstein, S. X. Xie, J. Y. Yee, R. M. Kotloff, D. A. Lipson and G. R. Bunin, Am. J. Respir. Crit. Care Med., 2006, 173, 659–666 CrossRef PubMed.
- (a) J. Costa, L. J. Benvenuto and J. R. Sonett, Best Pract. Res., Clin. Anaesthesiol., 2017, 31, 285–297 CrossRef PubMed; (b) S. E. Verleden, R. Vos, B. M. Vanaudenaerde and G. M. Verleden, J. Thorac. Dis., 2017, 9, 2650–2659 CrossRef PubMed.
- S. M. Kawut, Clin. Chest Med., 2011, 32, 367–377 CrossRef PubMed.
- Y. S. Prakash, D. J. Tschumperlin and K. R. Stenmark, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2015, 309, L625–L638 CrossRef CAS PubMed.
- R. Farre, J. Otero, I. Almendros and D. Navajas, Arch. Bronconeumol., 2018, 54, 31–38 Search PubMed.
- (a) J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS PubMed; (b) B. V. Slaughter, S. S. Khursi, O. Z. Fisher, A. Khademhosseini and N. A. Peppas, Adv. Mater., 2009, 21, 3307–3329 CrossRef CAS PubMed.
- (a) D. M. Faulk, S. A. Johnson, L. Zhang and S. F. Badylak, J. Cell. Physiol., 2014, 229, 984–989 CrossRef CAS PubMed; (b) S. Naahidi, M. Jafari, M. Logan, Y. Wang, Y. Yuan, H. Bae, B. Dixon and P. Chen, Biotechnol. Adv., 2017, 35, 530–544 CrossRef CAS PubMed.
- (a) G. Chen, J. Chen, Q. Liu, C. Ou and J. Gao, RSC Adv., 2015, 5, 30675–30678 RSC; (b) P. Li, Z. Yin, X.-Q. Dou, G. D. Zhou and C.-L. Feng, ACS Appl. Mater. Interfaces, 2014, 6, 7948–7952 CrossRef CAS PubMed; (c) R. S. Jacob, D. Ghosh, P. K. Singh, S. K. Basu, N. N. Jha, S. Das, P. K. Sukul, S. Patil, S. Sathaye, A. Kumar, A. Chowdhury, S. Malik, S. Sen and S. K. Maji, Biomaterials, 2015, 54, 97–105 CrossRef CAS PubMed; G. F. Liu, W. Ji, W. L. Wang and C. L. Feng, ACS Appl. Mater. Interfaces, 2015, 7, 301–307 Search PubMed.
- (a) R. G. Ellis-Behnke, Y.-X. Liang, S.-W. You, D. K. C. Tay, S. Zhang, K.-F. So and G. E. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5054–5059 CrossRef CAS PubMed; (b) R. G. Ellis-Behnke, G. Scheider and S. Zhang, US Pat., 7846891, 2010 Search PubMed; (c) G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I. Stupp, Science, 2004, 303, 1352–1355 CrossRef CAS PubMed; (d) J. D. Hartgerink, K. L. Niece and S. I. Stupp, WO2004106359, 2006; (e) V. M. Tysseling-Mattiace, V. Sahni, K. L. Niece, D. Birch, C. Czeisler, M. G. Fehlings, S. I. Stupp and J. A. Kessler, J. Neurosci., 2008, 28, 3814–3823 CrossRef CAS PubMed; (f) V. A. Kumar, N. L. Taylor, S. Shi, B. K. Wang, A. A. Jalan, M. K. Kang, N. C. Wickremasinghe and J. D. Hartgerink, ACS Nano, 2015, 9, 860–868 CrossRef CAS PubMed.
- R. Balint, N. J. Cassidy and S. H. Cartmel, Acta Biomater., 2014, 10, 2341–2353 CrossRef CAS PubMed.
- V. M. P. Vieira, L. L. Hay and D. K. Smith, Chem. Sci., 2017, 8, 6981–6990 RSC.
- (a) S. E. Sakiyama-Elbert and J. A. Hubbell, J. Controlled Release, 2000, 69, 149–158 CrossRef CAS; (b) J. A. Beamish, L. C. Geyer, N. A. Haq-Siddiqi, K. Kottke-Marchant and R. E. Marchant, Biomaterials, 2009, 30, 6286–6294 CrossRef CAS PubMed.
- B. O. Okesola, S. K. Suravaram, A. Parkin and D. K. Smith, Angew. Chem., Int. Ed., 2016, 55, 183–187 CrossRef CAS PubMed.
- H. Komatsu, S. Tsukiji, M. Ikeda and I. Hamachi, Chem. – Asian J., 2011, 6, 2368–2375 CrossRef CAS PubMed.
- (a) K. T. Nguyen and J. L. West, Biomaterials, 2002, 23, 4307–4314 CrossRef CAS PubMed; (b) N. Annabi, A. Tamayol, J. A. Uquillas, M. Akbari, L. E. Bertassoni, C. Cha, C. Camci-Unal, M. R. Dokmeci, N. A. Peppas and A. Khademhosseini, Adv. Mater., 2014, 36, 85–124 CrossRef; (c) E. Calo and V. V. Khutoryanskiy, Eur. Polym. J., 2015, 65, 252–267 CrossRef CAS.
- D. J. Cornwell, B. O. Okesola and D. K. Smith, Angew. Chem., Int. Ed., 2014, 53, 12461–12465 CAS.
- V. M. P. Vieira, A.-C. Lima, M. de Jong and D. K. Smith, unpublished results.
- D. K. Smith, Chemistry World, 2014, April, https://www.chemistryworld.com/opinion/no-sexuality-please-were-scientists/7197.article, accessed 05/02/18.
| This journal is © The Royal Society of Chemistry 2018 |