
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Negative dendritic effect on enzymatic hydrolysis of dendrimer conjugates†
Zhengwei
Zhou‡
a,
Mei
Cong‡
b,
Mengyao
Li
a,
Aura
Tintaru
c,
Jia
Li
d,
Jianhua
Yao
d,
Yi
Xia
 *a and
Ling
Peng
*b
*a and
Ling
Peng
*b
aChongqing Key Laboratory of Natural Product Synthesis and Drug Research, School of Pharmaceutical Sciences, Chongqing University, Chongqing, 401331, China. E-mail: yixia@cqu.edu.cn
bAix-Marseille Université, CNRS, Centre Interdisciplinaire de Nanoscience de Marseille, UMR 7325, “Equipe Labellisée Ligue Contre le Cancer”, Marseille 13288, France. E-mail: ling.peng@univ-amu.fr
cAix-Marseille Université, CNRS, UMR 7273, Institut de Chimie Radicalaire, Marseille, France
dShanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai, 200032, China
First published on 25th May 2018
Abstract
Dendrimers possess intriguing “dendritic effects”, which are unique characteristics that stem from the dendrimer generation and size. Here we report a “negative dendritic effect” observed during enzymatic hydrolysis of dendrimer conjugates. Such negative dendritic effects, though rarely reported, may be explored for tailored and generation-dependent drug release.
Drug delivery systems (DDSs) aim to improve drug bioavailability and facilitate controlled delivery of the drug to the disease lesion in order to maximize drug potency while minimizing toxicity.1–3 Dendrimers are emerging as promising DDSs because of their unique dendritic structure and multivalent cooperativity, which stem from their highly branched yet perfectly controllable architecture.4,5 Both physical encapsulation and chemical conjugation have been implemented for developing dendrimer-based DDSs in biomedical applications such as drug delivery, gene transfection, tissue engineering and imaging agents etc.6–13 We have been actively engaged in developing functional dendrimers as nanovectors for nucleic acid delivery,14,15 and we have recently established self-assembling amphiphilic dendrimer nanosystems to physically encapsulate drugs and nucleic acid molecules as innovative delivery platforms.16–19 Here, we report covalently conjugated amphiphilic dendrimers as drug delivery systems, with the aim of combining the advantages of multivalent dendrimers with stable drug loading. It is notable that, in addition to their well-known multivalent cooperativity, dendrimers also possess intriguing properties called “dendritic effects”, i.e. the unique characteristics of dendrimers which stem from and are associated with their different generation level and size.20,21 These dendritic effects are not only observed for the dendrimers themselves, but also for the functions grafted onto them, which lead to different properties.21,22 Remarkably, DDSs created from our amphiphilic dendrimer conjugates had “negative dendritic effects” on enzymatic hydrolysis for the release of bioactive entities. We expect that these negative dendritic effects, which are seldom reported, can be further exploited for constructing tailored and controllable DDSs.
For the dendrimer conjugates studied in this work, we chose the triazole nucleoside analog 1 as the bioactive drug candidate because of its potent anticancer activity and novel mode of action in inhibiting heat shock factor 1 (HSF1) and androgen receptor expression.23,24 In addition, nucleoside analogues are an important class of drug candidate and have been used in pharmacologically diverse applications, including as antiviral, anticancer, antimicrobial and immuno-modulatory agents.25 In the 2000s, Jacobson's group reported the use of dendrimer–nucleoside conjugates to improve the affinity and selectivity of adenosine derivatives as adenosine receptor agonists by harnessing the multivalent properties of dendrimers.26–28 Recently, Sk et al. exploited the steric crowdedness of dendrimer terminals to prevent rapid degradation of the nucleoside anticancer drug cytarabine (Ara-C) via covalent conjugation to PAMAM dendrimer terminals.29 Nevertheless, there is no report describing the conjugation of nucleoside analogues to amphiphilic dendrimers for drug delivery. In this work, we appended the nucleoside analogue 1 to the dendrimer terminals via biodegradable ester bonds with the aim of achieving drug release via enzymatic hydrolysis. Scheme 1A shows the enzymatic hydrolysis of a dendrimer conjugated with the nucleoside analogue 1, while Scheme 1B shows the two dendrimers I and II described in this work.
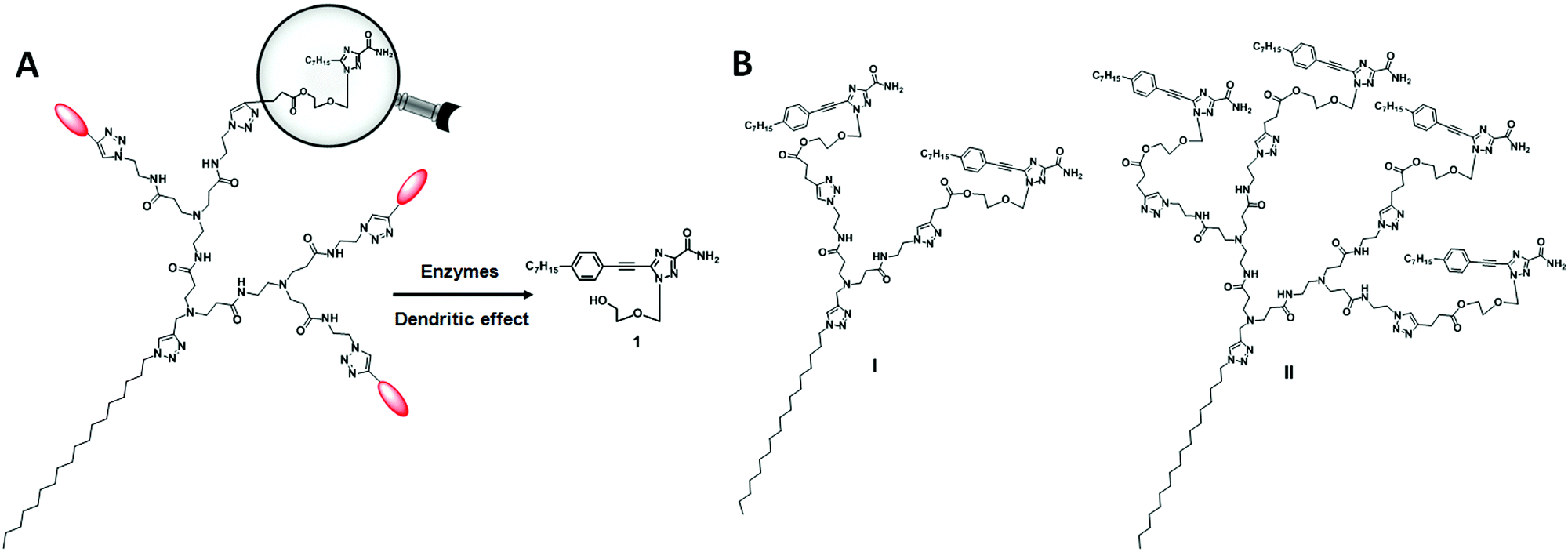 | ||
| Scheme 1 (A) Cartoon illustration of the enzymatic release of the biologically active parent nucleoside analogue from the covalent dendrimer conjugate. (B) Structures of the nucleoside–dendrimer conjugates I and II studied in this work. | ||
The synthesis of the dendrimer conjugates I and II was achieved successfully via click chemistry using the alkynyl- bearing nucleoside analogue 2 and the corresponding azido- containing dendrimers I-2 and II-2 (Scheme 2 and Schemes S1, S2, ESI†). 2 was made through esterification of 1 with 4-pentynoic acid, whereas I-2 and II-2 were prepared starting with the respective amine-terminated dendrimers I-1 and II-130via diazo-transfer.31 For the synthesis of dendrimers I and II, it should be mentioned that a slight excess of 2 considerably suppressed the formation of unknown side-products during the Cu(I)-catalyzed Huisgen click reaction.
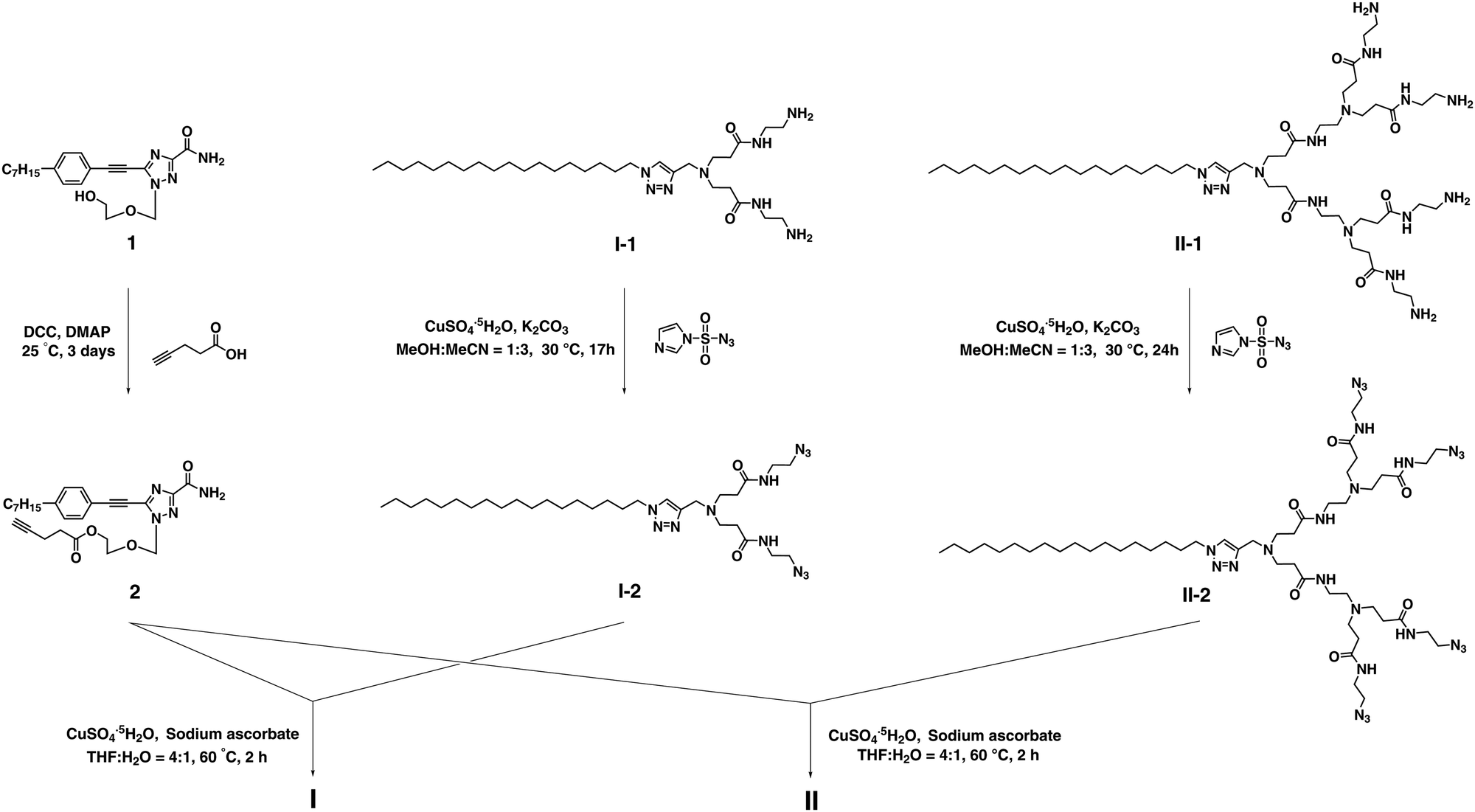 | ||
| Scheme 2 Synthesis of the nucleoside–dendrimer conjugates I and IIvia click chemistry. | ||
The chemical structure of both I and II was examined using 1H- and 13C-NMR. Fig. 1A illustrates the 1H-NMR spectra of II as an example. Compared to the precursor dendrimer II-2, the nucleoside–dendrimer conjugate II has new and characteristic 1H-NMR signals (Fig. 1A), which can be ascribed to the introduced 1,2,4-triazole nucleoside and the 1,2,3-triazole linkage. Likewise, the 13C-NMR spectrum of II (Fig. 1B) exhibits specific NMR signals belonging to the carbons in the triazole nucleoside component and the triazole connection, further supporting the chemical structures of I and II.
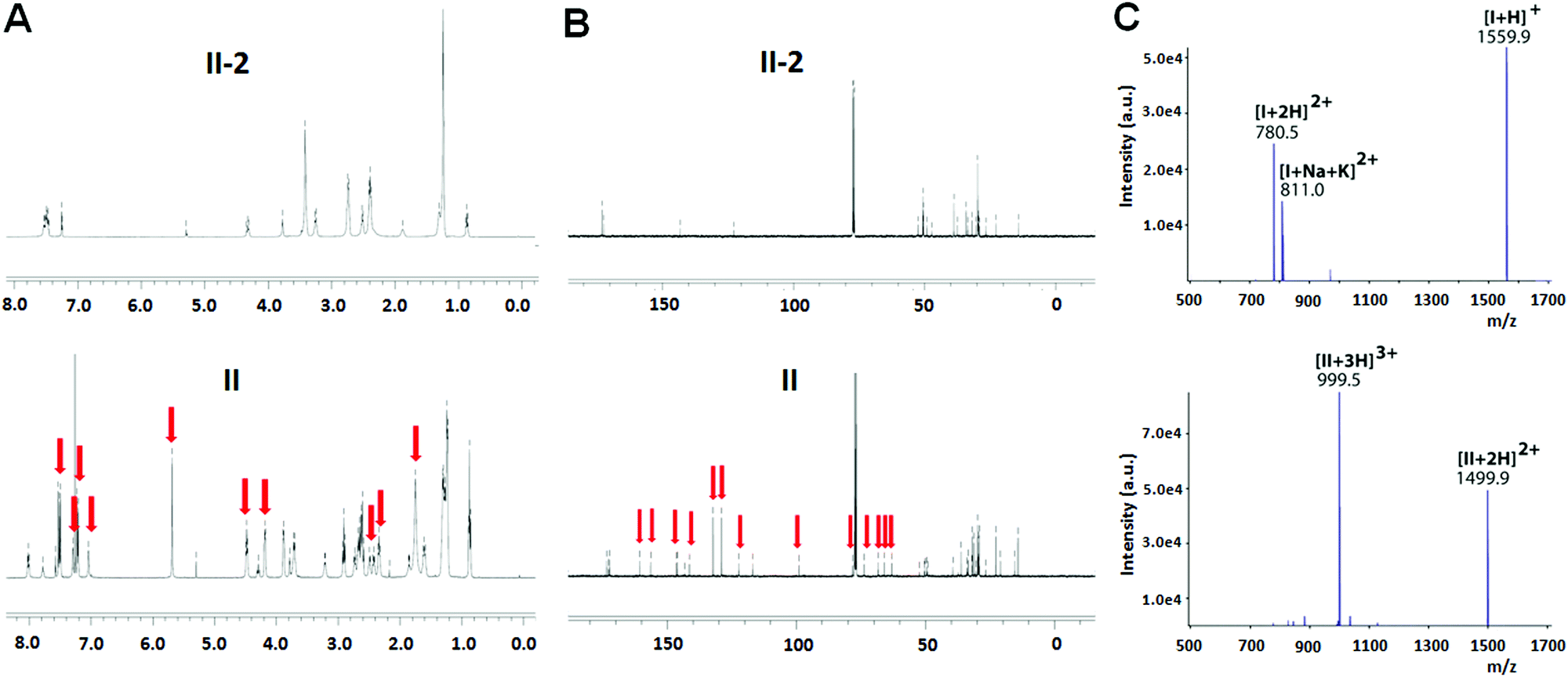 | ||
| Fig. 1 (A) 1H- and (B) 13C-NMR characterization of the triazole nucleoside–dendrimer conjugate II in comparison with the azide-terminated dendrimer II-2. The red arrows indicate the NMR signals related to the triazole nucleoside and linker entities in II. (C) ESI-MS spectra of the dendrimer conjugates I (top) and II (bottom). | ||
We also performed high-resolution mass spectrometry (MS) analysis of I and II. For I, the protonated species [I+H]+ was detected at m/z 1559.9 (Fig. 1C, top panel), while the ESI-MS spectrum of II (Fig. 1C, bottom panel) revealed the formation of double- and triple-protonated ions, observed at m/z 1499.9 and m/z 999.5, respectively. Further MS/MS analysis highlighted the characteristic structural features of the dendrimer conjugates (data not shown). Additionally, the elementary composition of the dendrimer conjugates I and II was confirmed by detection of molecular ions at the expected values: C83H122N20O102+ for I, ((m/z)th 780.4899, (m/z)exp 780.4899, error +0.0 ppm) and C155H225N40O223+ for II, ((m/z)th 999.5900, (m/z)exp 999.5902, error +0.2 ppm), respectively.
Both I and II are not soluble in water, with CMC values being 26 and 15 μM respectively. Nevertheless, they are readily soluble in the co-solvent systems of water and organic solvents. They are also stable at both physiological pH 7.4 and acidic pH 5.0, and no release of the nucleoside analogue 1 was observed even after 2 days.
We then studied the release of 1 from 2, I and II upon treatment with pig liver esterase (PLE) in fetal bovine serum (FBS) in the co-solvent system of DMSO/H2O (ESI†). PLE is widely applied to hydrolyze a wide variety of esters including water-insoluble esters, and can be used in co-solvent systems of H2O and organic solvents.32 Notably, the release of 1 from 2 was around 23% within 4 h, and this increased to 30% after 8 h (Fig. 2A). After 2 days, a total of 50% of 1 was released from 2. This result illustrates the effective enzymatic hydrolysis of the ester linkage in 2 to deliver 1. In contrast, the release of 1 from the dendrimer conjugate I was gradual and much slower than that from 2 (Fig. 2B). More remarkably, the release of 1 from the dendrimer conjugate II was dramatically reduced (Fig. 2C). Only around 10% of 1 was released after 2 days. The release rate of 1 from II is almost 2-fold slower than that from conjugate I, and around 5-fold slower than that from 2 at 4 h. This finding highlights that both I and II are much more resistant to enzymatic hydrolysis than 2 when releasing 1. This can be ascribed to the steric crowding of the dendrimer–nucleoside terminals, which impeded the access of the enzyme and hence the cleavage of the ester linkage. Importantly, this generation-dependent dendritic effect is a negative dendritic effect, unlike the synergic and enhanced effects that are usually associated with dendrimers.21
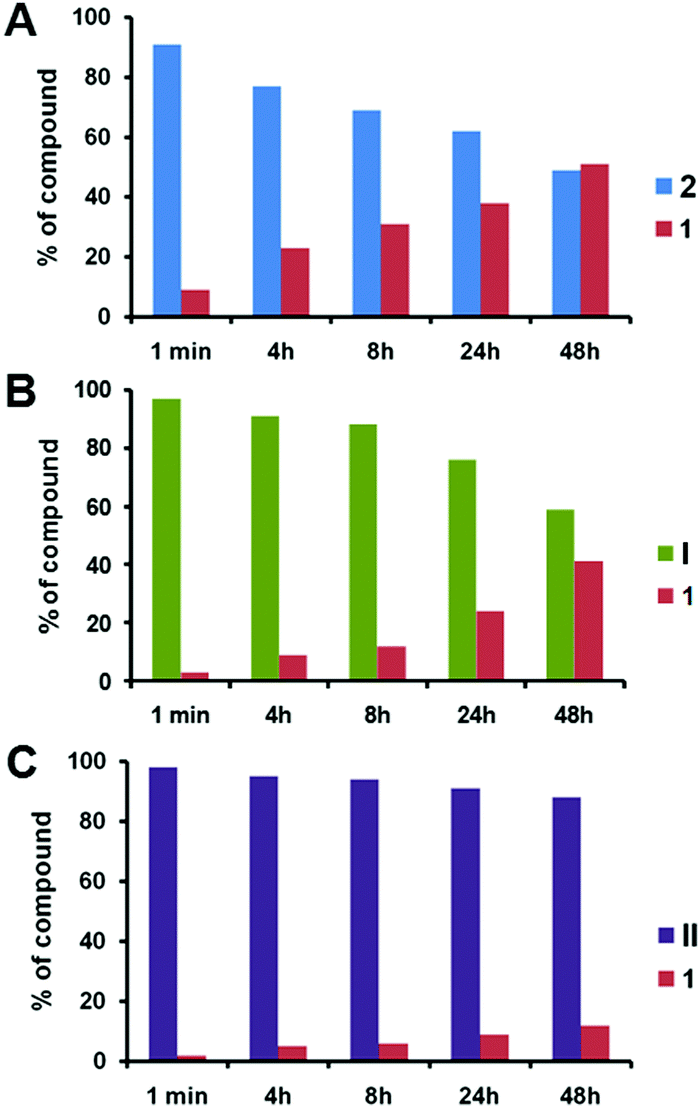 | ||
| Fig. 2 Enzymatic release of triazole nucleoside 1 from (A) 2, (B) I and (C) II in the presence of pig liver esterase in FBS. | ||
As bond energy is one important descriptor of bond stability, we further estimated the bond energy for the ester linkages in I, II and 2.33–35 The calculation results showed that the order of average bond energy of the ester linkage in 2, I and II was ΔE2 > ΔEI > ΔEII (Table S2, ESI†), indicating that the order of stability of the ester bond is 2 < I < II. Taken together, these data provide a reasonable explanation for the slower release of 1 from I and II than from 2, as well as the slower release from II than from I.
The negative dendritic effect is rarely reported in the literature and has been frequently ignored because it is often considered as uninteresting.21 Nevertheless, we think that it can be positively explored for drug delivery, because it can increase the stability of drug conjugates, hence allowing sustained drug release after accumulation of the conjugates in the disease lesion. For example, the covalent conjugation of the nucleoside anticancer drug cytarabine (Ara-C) to PAMAM dendrimer terminals prevented rapid metabolism and degradation of Ara-C.29 This can also be considered as a negative dendritic effect. The increased stability resulting from the negative dendritic effect can be tailored to design generation-dependent dendrimer conjugates for controlled release.
In summary, we successfully prepared covalent dendrimer conjugates using click chemistry and studied the release of a bioactive entity from the conjugates during enzymatic hydrolysis of the ester linkage. Our results showed that the enzymatic hydrolysis of the dendrimer conjugates was greatly impacted by dendritic structure and dendrimer generation. Higher-generation dendrimers had considerably slower enzymatic hydrolysis. This negative yet interesting dendritic effect can be positively exploited to design and construct generation-dependent dendrimer conjugates with tailored stability for achieving controllable drug release. We believe that covalent chemical conjugation is a promising alternative strategy to enable controlled delivery of therapeutic agents, and we are working actively in this direction.
Financial support was from the National Natural Science Foundation of China (81502920, 21672251), the Chongqing Natural Science Foundation (cstc2015jcyjA10093), the Fundamental Research Funds for the Central Universities (106112017CDJQJ468823), the Instrumental Analysis Foundation of Chongqing University (201712150020), La Ligue Nationale Contre le Cancer (EL2016.LNCC/LPP) and the French National Research Agency under the frame of EuroNanoMed II (ANR-15-ENM2-0006-02, ANR-16-ENM2-0004-02). We thank Prof. Zhining Xia for HPLC assistance.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Mitragotri and J. Lahann, Adv. Mater., 2012, 24, 3717–3723 CrossRef PubMed.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2014, 13, 813–827 CrossRef PubMed.
- G. Chen, I. Roy, C. Yang and P. N. Prasad, Chem. Rev., 2016, 116, 2826–2885 CrossRef PubMed.
- Dendrimers, Dendrons, and Dendritic Polymers: Discovery, Applications, and the Future, ed. D. A. Tomalia, J. B. Christensen and U. Boas, Cambridge University Press, 2012 Search PubMed.
- D. A. Tomalia, A. M. Naylor and W. A. Goddard III, Angew. Chem., Int. Ed. Engl., 1990, 29, 138–175 CrossRef.
- C. C. Lee, J. A. MacKay, J. M. J. Fréchet and F. C. Szoka, Nat. Biotechnol., 2005, 23, 1517–1526 CrossRef PubMed.
- M. A. Mintzer and M. W. Grinstaff, Chem. Soc. Rev., 2011, 40, 173–190 RSC.
- S. Svenson and D. A. Tomalia, Adv. Drug Delivery Rev., 2005, 57, 2106–2129 CrossRef PubMed.
- Dendrimers in Biomedical Applications, ed. B. Klajnert, L. Peng and V. Cena, Royal Society of Chemistry, 2013 Search PubMed.
- J. Yang, Q. Zhang, H. Chang and Y. Cheng, Chem. Rev., 2015, 115, 5274–5300 CrossRef PubMed.
- B. Gorain, M. Tekade, P. Kesharwani, A. K. Iyer, K. Kalia and R. K. Tekade, Drug Discovery Today, 2017, 22, 652–664 CrossRef PubMed.
- S. H. Medina and M. E. H. El-Sayed, Chem. Rev., 2009, 109, 3141–3157 CrossRef PubMed.
- D. Astruc, E. Boisselier and C. Ornelas, Chem. Rev., 2010, 110, 1857–1959 CrossRef PubMed.
- J. Zhou, J. Wu, N. Hafdi, J. P. Behr, P. Erbacher and L. Peng, Chem. Commun., 2006, 2362–2364 RSC.
- J. Ma, S. Kala, S. Yung, T. M. Chan, Y. Cao, Y. Jiang, X. Liu, S. Giorgio, L. Peng and A. S. T. Wong, Mol. Ther., 2018, 26, 70–83 CrossRef PubMed.
- T. Wei, C. Chen, J. Liu, C. Liu, P. Posocco, X. Liu, Q. Cheng, S. Huo, Z. Liang, M. Fermeglia, S. Pricl, X. J. Liang, P. Rocchi and L. Peng, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2978–2983 CrossRef PubMed.
- X. Liu, J. Zhou, T. Yu, C. Chen, Q. Cheng, K. Sengupta, Y. Huang, H. Li, C. Liu, Y. Wang, P. Posocco, M. Wang, Q. Cui, S. Giorgio, M. Fermeglia, F. Qu, S. Pricl, Y. Shi, Z. Liang, P. Rocchi, J. J. Rossi and L. Peng, Angew. Chem., Int. Ed. Engl., 2014, 53, 11822–11827 CrossRef PubMed.
- X. Liu, Y. Wang, C. Chen, A. Tintaru, Y. Cao, J. Liu, F. Ziarelli, J. Tang, H. Guo, R. Rosas, S. Giorgio, L. Charles, P. Rocchi and L. Peng, Adv. Funct. Mater., 2016, 26, 8594–8603 CrossRef.
- C. Chen, P. Posocco, X. Liu, Q. Cheng, E. Laurini, J. Zhou, C. Liu, Y. Wang, J. Tang, V. D. Col, T. Yu, S. Giorgio, M. Fermeglia, F. Qu, Z. Liang, J. J. Rossi, M. Liu, P. Rocchi, S. Pricl and L. Peng, Small, 2016, 12, 3667–3676 CrossRef PubMed.
- D. A. Tomalia, New J. Chem., 2012, 36, 264–281 RSC.
- A. M. Caminade, A. Ouali, R. Laurent, C. O. Turrin and J. P. Majoral, Chem. Soc. Rev., 2015, 44, 3890–3899 RSC.
- G. Zaupa, P. Scrimin and L. J. Prins, J. Am. Chem. Soc., 2008, 130, 5699–5709 CrossRef PubMed.
- Y. Xia, M. Wang, E. Beraldi, M. Cong, A. Zoubeidi, M. Gleave and L. Peng, Anticancer Agents Med. Chem., 2015, 15, 1333–1340 CrossRef PubMed.
- Y. Xia, Y. Liu, P. Rocchi, M. Wang, Y. Fan, F. Qu, J. L. Iovanna and L. Peng, Cancer Lett., 2012, 318, 145–153 CrossRef PubMed.
- L. P. Jordheim, D. Durantel, F. Zoulim and C. Dumontet, Nat. Rev. Drug Discovery, 2013, 12, 447–464 CrossRef PubMed.
- A. M. Klutz, Z. G. Gao, J. Lloyd, A. Shainberg and K. A. Jacobson, J. Nanobiotechnol., 2008, 6, 12 CrossRef PubMed.
- D. K. Tosh, L. S. Yoo, M. Chinn, K. Hong, S. M. Kilbey II, M. O. Barrett, I. P. Fricks, T. K. Harden, Z. G. Gao and K. A. Jacobson, Bioconjugate Chem., 2010, 21, 372–384 CrossRef PubMed.
- D. K. Tosh, K. Phan, F. Deflorian, Q. Wei, L. S. Yoo, Z. G. Gao and K. A. Jacobson, Bioconjugate Chem., 2012, 23, 232–247 CrossRef PubMed.
- U. H. Sk, S. P. Kambhampati, M. K. Mishra, W. G. Lesniak, F. Zhang and R. M. Kannan, Biomacromolecules, 2013, 14, 801–810 CrossRef PubMed.
- T. Yu, X. Liu, A. L. Bolcato-Bellemin, Y. Wang, C. Liu, P. Erbacher, F. Qu, P. Rocchi, J. P. Behr and L. Peng, Angew. Chem., Int. Ed. Engl., 2012, 51, 8478–8484 CrossRef PubMed.
- K. Barral, A. D. Moorhouse and J. E. Moses, Org. Lett., 2007, 9, 1809–1811 CrossRef PubMed.
- Enzymes in Synthetic Organic Chemistry, ed. C. H. Wong and G. M. Whitesides, Pergamon, 1994 Search PubMed.
- M. J. S. Dewar, E. G. Zoebisch, E. F. Healy and J. J. P. Stewart, J. Am. Chem. Soc., 1985, 107, 3902–3909 CrossRef.
- HyperChem Pro 7.5 for Windows and NT. Hypercube Inc.
- K. Morokuma, J. Chem. Phys., 1971, 55, 1236–1244 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc01221k |
| ‡ ZZ and MC contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |