
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atomic-level design of CoOH+–hydroxyapatite@C catalysts for superfast degradation of organics via peroxymonosulfate activation†
Feng
Song
a,
Huayang
Zhang
 b,
Shaobin
Wang
*b,
Lihong
Liu
*b,
Xiaoyao
Tan
c and
Shaomin
Liu
b
b,
Shaobin
Wang
*b,
Lihong
Liu
*b,
Xiaoyao
Tan
c and
Shaomin
Liu
b
aSchool of Chemical Engineering, Shandong University of Technology, China
bDepartment of Chemical Engineering, Curtin University, WA 6845, Australia. E-mail: Shaobin.wang@curtin.edu.au
cSchool of Environmental and Chemical Engineering, Tianjin Polytechnic University, China
First published on 30th March 2018
Abstract
We report a strategy for simultaneous cobalt removal and organic waste decomposition by using mesoporous hydroxyapatite nanoparticles wrapped in uniform carbon layers (HA@C). The in situ formation of CoOH+–HA@C due to ion exchange greatly improved the degradation efficiency by at least one order of magnitude compared to free Co2+.
Toxic heavy metal ions discharged mostly from the foundry and mining industries have underlined the high importance of wastewater treatment. Recent technologies for heavy metal elimination include chemical precipitation, adsorption, ion-exchange, membrane filtration and electrochemical deposition.1,2 Among them, adsorption using certain solids to capture specific ions from solutions is widely accepted due to easy operation. For example, hydroxyapatite (HA), with an empirical formula of Ca10(PO4)6(OH)2, has been extensively studied to immobilize many divalent heavy metal ions in wastewater and even in the human body. The stabilization mechanisms of Pb2+, Zn2+, Co2+, Cd2+, Cu2+, Hg2+ and Ni2+ on HA involve dissolution–precipitation, ion-exchange, surface complexation and coprecipitation.3–6 Although previous techniques work well for simple heavy metal removal, approaches for in situ reuse of the captured metal ions are very limited.
Traditional methods for the synthesis of HA nanoparticles are solid-state reactions, precipitation, sol–gel techniques, electrospinning synthesis, electrodeposition, hydrothermal reactions, and so on.7–12 In the past few decades, advancements in biotechnology have led to many sustainable ways to prepare nanomaterials with plant extracts or microorganisms.13,14 Several studies have shown that Serratia sp. bacteria derived HA exhibited superior sorption for the removal of Sr2+ and Co2+.15 Recently, our research group has simplified the microbial process by using Yeast Mold Broth (YMB) to implement fine control over the carbon- and silver-modified hierarchical ZnO formation.16–18 YMB powder has 47.6% dextrose (D-glucose) that can be used as a carbon source. In the present study, we report the synthesis of HA nanorods (HA@C) with a mesoporous core and a uniform carbon shell structure. The HA@C nanocomposite could effectively remove Co2+ from aqueous solution, for instance 83% Co2+ removal efficiency from 6 ppm aqueous solution. To make the wastewater treatment process more economical, we propose to use the captured Co2+ for further advanced oxidation of organic pollutants via peroxymonosulfate (PMS) decomposition and the subsequently produced more potent radicals.19,20 The detailed mechanism of PMS activation by cobalt is generally proposed as follows.21–24
| Co2+ + H2O → CoOH+ + H+ | (1) |
| CoOH+ + HSO5− → CoO+ + SO4˙− + H2O | (2) |
| SO4˙− + organics → intermediate products → CO2 + H2O | (3) |
CoOH+ is one of the most effective species to activate PMS. Thus, the formation of CoOH+ is considered as the rate-limiting step in the process. To facilitate the generation of Co–OH complexes, Yang et al. immobilized Co3O4 onto TiO2 nanoparticles.25 The obtained Co/TiO2 catalyst demonstrated greatly enhanced heterogeneous PMS activation, due to the ability of TiO2 to dissociate H2O for surface hydroxyl group generation.
The HA crystal consists of hydroxyl groups positioned in the channel along the (001) direction.26 Therefore, in Co2+ adsorption, Ca2+ replacement by Co2+ could result in ready formation of CoOH+, which has a high potential function as the PMS catalytic active center. Our experimental results indicate that Co2+ substituted HA@C/PMS ([Co2+] = 1 ppm) completely decomposed methylene blue in 1 min, while it took 45 min in a homogeneous Co2+/PMS system. Electron paramagnetic resonance (EPR) and quenching studies reveal that SO4˙−, ˙OH and 1O2 participated in the advanced oxidation process.
X-ray diffraction (XRD) patterns of the synthesized samples are presented in Fig. S1a (ESI†). At room temperature, bare HA particles with poor crystallinity were formed after 20 h. It is clear that the sample 4 × 180 °C 5 h HA (with a molar ratio of KH2PO4/dextrose = 4, hydrothermal treatment at 180 °C for 5 h) has intense diffraction peaks that match well with the standard data of JCPDS Card No. 09-0432. The sharp peaks located at 2θ = 26.0, 31.8, 32.2, 32.9, 39.9, and 46.7° can be indexed to the (002), (211), (112), (300), (310) and (222) planes of HA, respectively.27–29 All the samples prepared at 120 °C have similar characteristic peaks, though in relatively broader widths. The XRD patterns confirmed that predominately HA rather than other calcium phosphates has been obtained.30 Fig. S1b (ESI†) shows the absorption bands at ca. 560, 599 and 960 cm−1 corresponding to the vibration of PO43− groups in HA.27,30 The IR peak for the hydroxyl groups of HA is observed at around 628 cm−1. For X-ray photoelectron spectroscopy (XPS) characterization, two peaks with binding energies of 437 and 346 eV are in agreement with the characteristics of Ca 2s and Ca 2p. The peaks near 190 and 132 eV are attributed to P 2s and P 2p, respectively.29 The two peaks near 346 and 350 eV are attributed to the Ca 2p3/2 and Ca 2p1/2 bands of HA. The O 1s peaks near 530 and 532 eV are associated with the phosphate group in HA and adsorbed water, respectively.
Fig. 1a shows the typical rod-like morphology of 4 × 180 °C 5 h HA nanoparticles with numerous mesopores around 2–3 nm. It can be seen that the nanorod is approximately 20–30 nm in width, with a length varying between 30 and 100 nm. The interplanar spacings of ca. 0.81, 0.27 and 0.34 nm obtained from the HRTEM images were ascribed to the adjacent (100), (112) and (002) planes of HA NPs, respectively.30–34 Additionally, the particles are found to be covered with a uniform layer of amorphous carbon. The thickness of the carbon shell is about 10 nm (Fig. 1c). Important advantages of this structure over bare HA NPs are stabilization and introduction of a C source for PMS activation, which will be discussed later.
 | ||
| Fig. 1 TEM micrographs of hydroxyapatite (HA) nanorods wrapped with uniform carbon layers (a and c) and HRTEM images of HA crystals showing different lattice fringes (b and d). | ||
Zeta potential measurements of bare HA particles had a range around −36 to −25 mV.35,36 The surface charge of HA@C was approximately −11 mV in this study. The carbon layer may interfere with the OH− in attracting counter ions thus leading to the variation. HA@C NPs were observed to settle gradually over the time course of hours. In terms of operation cost reduction, HA@C may be preferred over highly stable HA NPs in avoiding additional catalyst separation, e.g., centrifugation. Nevertheless, they can be re-dispersed well by just hand shaking and show a high capacity in adsorption of cationic ions.
Fig. S2 (ESI†) depicts the N2 adsorption–desorption isotherm and the Barrett–Joyner–Halenda (BJH) pore size distribution of HA@C NPs. The isotherm is of type IV with a steep H3 hysteresis loop occurring in the range of p/p0 = 0.8–1.0, indicating the existence of irregular mesopores. The specific surface area, adsorption cumulative pore volume, and average pore diameter were found to be 88 m2 g−1, 0.41 cm3 g−1, and 18.8 nm, respectively. These values are comparable to previous studies.37
The adsorption and catalytic performance of HA@C NPs are shown in Fig. 2a. PMS itself oxidized 30% MB within the examined time. The HA adsorption capacities of MB and Co2+ are 3.5 and 5.0 mg g−1, respectively. At an initial concentration of 5–6 ppm, the Co2+ uptake was almost 10 times higher than multiwalled carbon nanotubes.38 When Co2+ was mixed with HA@C first, only 27% of MB was removed due to the competition of cationic metal ions and dye molecules for adsorption sites on HA@C.
 | ||
| Fig. 2 Methylene blue (MB) adsorption and degradation by PMS, HA@C/PMS and Co2+-loaded HA@C/PMS (a). Effect of Co2+ concentration on the homogeneous MB degradation process (b). The time for 100% MB removal and leached [Co2+] after multiple use of CoHA@C (1 g L−1) (c). [MB] = 10 ppm, 1 g L−1 HA@C, 0.1, 0.2, and 1.0 g L−1 CoHA@C (corresponding to [Co2+] of 0.5, 1, and 5 ppm, respectively), 0.24 g L−1 PMS (0.4 mM), pH 5.5, 25 °C. | ||
Metal-free carbon/PMS have been investigated for pollutant degradation. The sp2 carbons and O-containing groups on nanocarbons have demonstrated high efficiency in activating HSO5− for sulfate radical production.39–42 In this study, HA@C/PMS gradually degraded 85% MB, which was higher than HA@C adsorption (37%). This suggests that the amorphous carbon layer could possibly activate PMS for MB decomposition.
After Co2+ loading, CoHA@C (0.2 g L−1) could activate PMS and completely decolorized MB in 1 min. The system maintained the similar performance from pH 5 to pH 6.5 (Fig. S3, ESI†). This rate was even faster than the homogeneous process, as it took 45, 10 and 6 min for 1, 4 and 6 ppm free Co2+ to remove MB, respectively. To exclude the possibility of MB degradation by leached Co2+, we measured the [Co2+] in the supernatant using ICP-OES. Fig. 2c shows free [Co2+] decreasing from 0.8 to 0.4 ppm after the fourth reaction. Catalyst reusability tests were performed by conducting 4 rounds of the reaction. Although it took longer time, 100% MB removal could be achieved within 2 min in the fourth cycle. The decline in the degradation rate may be caused by the consumption of Co2+ attached on the carbon layer after the first reaction. However, the surface negative HA core has attracted and accommodated well to Co2+ for continuous PMS activation.
In most cases, SO4˙− is accepted as the major oxidizing agent in Co/PMS AOPs. In order to identify the dominating oxidizing species responsible for MB decontamination, EPR experiments were performed to detect the reactive oxygen species (ROSs) during PMS activation. As shown in Fig. 3a, 1 g L−1 CoHA@C, 6 ppm free Co2+ and PMS control produced stronger DMPO–˙OH signals (consisting of a quartet with an intensity ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1) than SO4˙− signals (marked with +).
:2:2:1) than SO4˙− signals (marked with +).
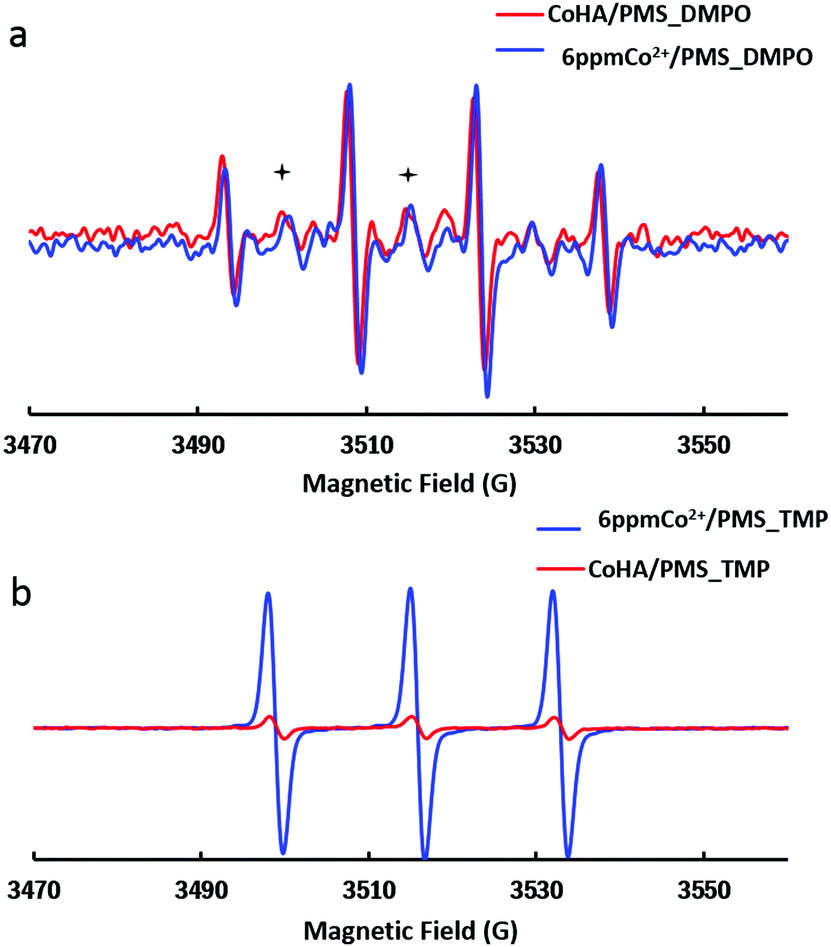 | ||
| Fig. 3 EPR tests on SO4˙−, ˙OH (a) and 1O2 (b). 1 g L−1 CoHA@C, 0.24 g L−1 PMS, pH 5.5, 25 °C. | ||
Another chemical, 2,2,6,6-tetramethyl-4-piperidinol (TMP), was used as a probe to confirm the generation of singlet oxygen. The EPR spectrum of the nitroxide radical (three equal intensity lines) indicated that 1O2 converts TMP to TMPO in a 6 ppm Co2+/PMS system. Again, only a trace level of 1O2 was detected in the CoHA@C/PMS system.
Ethanol is a widely used scavenger of SO4˙− and ˙OH.43 To further verify the existence of the radicals, quenching experiments were conducted by adding ethanol (ethanol:PMS = 1000) and NaN3 (3 mM) into the suspension for more efficient quenching of radicals. The degradation rate was notably inhibited as no MB decolorization was observed within 10 min, thus proving that the sulfate radical, hydroxyl radical and singlet oxygen indeed play key roles in the CoHA@C/PMS oxidation.
Considering the crystalline structure of HA, a mechanism for MB decomposition in the CoHA@C/PMS system was proposed as follows. Co2+ adsorption occurs due to ion exchange with Ca2+ in the surface groups of HA and also within the HA crystals. As depicted in Fig. 4, between the two Ca sites in the unit cell of crystalline HA, where metal ion exchange may happen, the Ca-2 site is normally replaced by Co2+.44 The Co2+ reacts with the adjacent OH− to form CoOH+, resulting in the most efficient catalyst for PMS decomposition. Additionally, an array of PO4 tetrahedra may act as the nucleophile (Nu) polyphosphate in PMS self-decomposition to generate 1O2.45
| HSO5− + Nu → NuOH+ + SO42− | (4) |
| HSO5− + NuOH+ → SO42−/HSO4− + 1O2 + Nu + H+ | (5) |
| 1O2 + methylene blue → intermediates + CO2 + H2O | (6) |
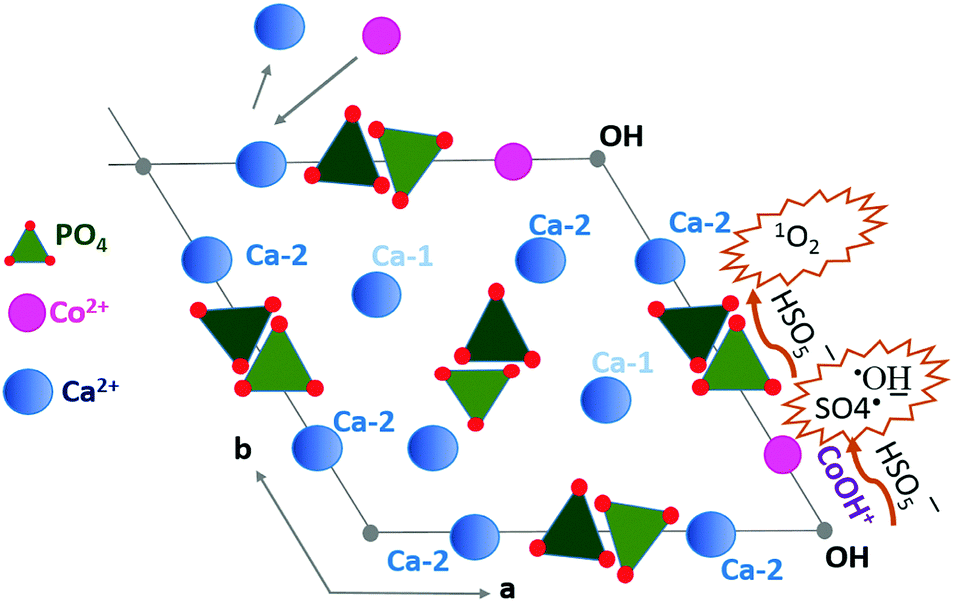 | ||
| Fig. 4 Proposed mechanism of Co2+ adsorption and radical production facilitated by CoOH+ and the nucleophile tetrahedral PO4. | ||
In EPR characterizations, 6 ppm free Co2+ produced stronger radical intensities, but with less efficiency than that of 5 ppm CoHA@C. This could be explained by the radical confinement inside the nanopores of mesoporous HA. Wang et al. prepared nitrogen-doped reduced graphene oxide and investigated its bisphenol elimination efficiency.46 By employing methanol and KI as radical scavengers, they confirmed that the surface-bound SO4˙− played a dominant role and only a minimal amount of free SO4˙− was generated in the AOP. Similarly, in our system, the adsorbed MB molecules were firstly degraded by various confined radicals. After releasing the occupied sites, a new adsorption–degradation of MB started again until all MB was decomposed.
In summary, a facile method is established for the bioinspired synthesis of hydroxyapatite nanorods with mesoporous structures and uniform carbon shells. The resultant HA@C composite is stable and efficient in adsorbing Co2+ from wastewater. At [Co] = 1.0 ppm, the in situ formed Co–OH complexes greatly reduce the dye degradation time by a factor of 45 compared to the conventional PMS activation by free Co2+. The SO4˙−, ˙OH and 1O2 are experimentally confirmed to play a vital role in the AOPs. The ultrafast decontamination rate and excellent reusability make the system a promising candidate for sustainable wastewater treatment.
Conflicts of interest
There are no conflicts to declare.Notes and references
- F. Fu and Q. Wang, J. Environ. Manage., 2011, 92, 407 CrossRef CAS PubMed
.
- A. Azimi, A. Azari, M. Rezakazemi and M. Ansarpour, ChemBioEng Rev., 2017, 4, 37 CrossRef CAS
- M. Vila, S. Sánchez-Salcedo and M. Vallet-Regí, Inorg. Chim. Acta, 2012, 393, 24 CrossRef CAS
- M. Vila, S. Sánchez-Salcedo, M. Cicuéndez, I. Izquierdo-Barba and M. Vallet-Regí, J. Hazard. Mater., 2011, 192, 71 CAS
- E. Mavropoulos, A. Malta Rossi and A. M. Costa, Environ. Sci. Technol., 2002, 36, 1625 CrossRef CAS PubMed
- G. Liu, J. W. Talley, C. Na, S. L. Larson and L. G. Wolfe, Environ. Sci. Technol., 2010, 44, 1366 CrossRef CAS PubMed
- M. Okada and T. Furuzono, Sci. Technol. Adv. Mater., 2012, 13, 064103 CrossRef PubMed
- H. Peng, J. Wang, S. Lv, J. Wen and J. Chen, Ceram. Int., 2015, 41, 14340 CrossRef CAS
- J. Chen, Y. Wang, X. Chen, L. Ren, C. Lai, W. He and Q. Zhang, Mater. Lett., 2011, 65, 1923 CrossRef CAS
- Y. Wu, L. L. Hench, J. Du, K. Choy and J. Guo, J. Am. Ceram. Soc., 2004, 87, 1988 CrossRef CAS
- A. Kar, K. S. Raja and M. Misra, Surf. Coat. Int., 2006, 201, 3723 CrossRef CAS
- Y. Yang, Q. Wu, M. Wang, J. Long, Z. Mao and X. Chen, Cryst. Growth Des., 2014, 14, 4864 CAS
- C. L. Keat, A. Aziz, A. M. Eid and N. A. Elmarzugi, Bioresour. Bioprocess., 2015, 2, 47 CrossRef
- Y. Park, Toxicol. Res., 2014, 30, 169 CrossRef CAS PubMed
- S. Handley-Sidhu, T. K. Mullan, Q. Grail, M. Albadarneh, T. Ohnuki and L. E. Macaskie, Sci. Rep., 2016, 6, 23361 CrossRef CAS PubMed
- Z. Shen, B. Liu, V. Pareek, S. Wang, X. Li, L. Liu and S. Liu, RSC Adv., 2015, 5, 80488 RSC
- S. Zhang, H. Zhang, S. Wang, L. Liu and S. Liu, Catal. Sci. Technol., 2017, 7, 4355 CAS
- Z. Shen, P. Liang, S. Wang, L. Liu and S. Liu, ACS Sustainable Chem. Eng., 2015, 3, 1010 CrossRef CAS
- G. Boczkaj and A. Fernandes, Chem. Eng. J., 2017, 320, 608 CrossRef CAS
- M. Gągola, A. Przyjazny and G. Boczkaj, Chem. Eng. J., 2018, 338, 599 CrossRef
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2003, 37, 4790 CrossRef CAS PubMed
- P. Hu and M. Long, Appl. Catal., B, 2016, 181, 103–117 CrossRef CAS
- R. Luo, C. Liu, J. Li, C. Wang, X. Sun, J. Shen, W. Han and L. Wang, J. Mater. Chem. A, 2018, 6, 3454 CAS
- C. Wang, H. Wang, R. Luo, C. Liu, J. Li, X. Sun, J. Shen, W. Han and L. Wang, Chem. Eng. J., 2017, 330, 262 CrossRef CAS
- Q. Yang, H. Choi and D. D. Dionysiou, Appl. Catal., B, 2007, 74, 170 CrossRef CAS
- N. H. de Leeuw, Chem. Commun., 2001, 1646 RSC
- W. Fang, H. Zhang, J. Yin, B. Yang, Y. Zhang, J. Li and F. Yao, Cryst. Growth Des., 2016, 16, 1247 CAS
- Y. K. Liu, D. D. Hou and G. H. Wang, Mater. Chem. Phys., 2004, 86, 69 CrossRef CAS
- M. Selvakumar, P. S. Kumar, B. Das, S. Dhara and S. Chattopadhyay, Cryst. Growth Des., 2017, 17, 433 CAS
- Y. Wang, X. Ren, X. Ma, W. Su, Y. Zhang, X. Sun and X. Li, Cryst. Growth Des., 2015, 15, 1949 CAS
- Y. Yang, Q. Wu, M. Wang, J. Long, Z. Mao and X. Chen, Cryst. Growth Des., 2014, 14, 4864 CAS
- H. Pan, X. Liu, R. Tang and H. Xu, Chem. Commun., 2010, 46, 7415 RSC
- K. A. Selvig, Calcif. Tissue Res., 1970, 6, 227 CrossRef CAS PubMed
- H. Zhang, M. Liu, H. Fan and X. Zhang, Cryst. Growth Des., 2012, 12, 2204 CAS
- D. Li, X. Huang, Y. Wu, J. Li, W. Cheng, J. He, H. Tian and Y. Huang, Biomater. Sci., 2016, 4, 272 RSC
- C. M. Botelho, M. A. Lopes, I. R. Gibson, S. M. Best and J. D. Santos, J. Mater. Sci.: Mater. Med., 2002, 13, 1123 CrossRef CAS PubMed
- H. Zhou, M. Yang, S. Hou and L. Deng, Mater. Sci. Eng., C, 2017, 71, 439 CrossRef CAS PubMed
- A. Stafiej and K. Pyrzynska, Sep. Purif. Technol., 2007, 58, 49 CrossRef CAS
- S. Yang, T. Xiao, J. Zhang, Y. Chen and L. Li, Sep. Purif. Technol., 2015, 143, 19 CrossRef CAS
- E. Saputra, S. Muhammad, H. Sun and S. Wang, RSC Adv., 2013, 3, 21905 RSC
- H. Sun, C. K. Kwan, A. Suvorova, H. M. Ang, M. O. Tadé and S. Wang, Appl. Catal., B, 2014, 154, 134 CrossRef
- H. Sun, S. Liu, G. Zhou, H. M. Ang, M. O. Tadé and S. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 5466 CAS
- W. Tian, H. Zhanga, Z. Qian, T. Ouyang, H. Sun, J. Qin, M. Tadé and S. Wang, Appl. Catal., B, 2018, 225, 76 CrossRef CAS
- M. E. Zilm, L. Chen, V. Sharma, A. McDannald, M. Jain, R. Ramprasad and M. Wei, Phys. Chem. Chem. Phys., 2016, 18, 16457 RSC
- X. Lou, C. Fang, Z. Geng, Y. Jin, D. Xiao, Z. Wang, J. Liu and Y. Guo, Chemosphere, 2017, 173, 529 CrossRef CAS PubMed
- X. Wang, Y. Qin, L. Zhu and H. Tang, Environ. Sci. Technol., 2015, 49, 6855 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc00946e |
| This journal is © The Royal Society of Chemistry 2018 |