
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of charge in 1,2,3-triazol(ium)-based halogen bonding activators†
Alexander
Dreger
a,
Elric
Engelage
a,
Bert
Mallick
a,
Paul D.
Beer
b and
Stefan M.
Huber
 *a
*a
aDepartment of Chemistry and Biochemistry, Ruhr-Universität Bochum, Universitätsstraße 150, 44801 Bochum, Germany. E-mail: stefan.m.huber@rub.de
bDepartment of Chemistry, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK
First published on 4th April 2018
Abstract
The influence of charge on the performance of monocationic and dicationic triazol(ium)-based halogen bond donors was investigated. Next to the activity in a halide abstraction benchmark reaction, halogen bonding was also evaluated via X-ray structural analyses and isothermal titration calorimetry.
Halogen bonds (XBs) are highly directional non-covalent interactions between the electrophilic region of a covalently bound halogen atom and a Lewis base.1 For strong halogen bonding, the respective XB donor (i.e., the halogen-based Lewis acid) requires an electron-withdrawing core structure, and often perfluorinated or cationic (hetero)arenes are used. Until now, halogen bonding has been successfully applied in many areas, including crystal engineering, supramolecular chemistry and organocatalysis.2–4
Halogen bond donors based on triazolium moieties were first introduced in the context of an anion-binding rotaxane.5 Bidentate triazolium-based activators were then used in a benchmark reaction for halide abstraction (the solvolysis of benzhydryl bromide 1 in acetonitrile), and their activating power was found6 to be similar to that of bidentate imidazolium7 or pyridinium-based8 halogen bond donors. Subsequently, neutral bidentate triazole derivatives were investigated as anion receptors,9,10 but their binding strength to chloride was found to be markedly lower than that of their dicationic analogues. This is in line with previous findings that charge-assisted XB donors are stronger Lewis acids11 and are thus preferentially used in organic synthesis. On the other hand, halogen bond donors based on cationic core structures also feature some intrinsic disadvantages, e.g. low solubility in apolar solvents and competition by Lewis basic counterions.12
In 2015, Beer et al. introduced strong bidentate (but monocationic) halogen bond donors based on a bis(iodotriazolyl)pyridinium motif (3,5-Py(X)R,R′/Z, Fig. 1 middle) for anion recognition.13 Since these compounds represent an interesting compromise between overall charge assistance but neutral halogen-bond-donating moieties, we became interested to test their performance as Lewis acidic activators, especially in comparison to our previous dicationic systems (1,3-Ph(I)Oct,Me/OTf, Fig. 1 left).6 In doing so, our main goal was to elucidate the relevance of charge on the actual halogen bonding groups.14
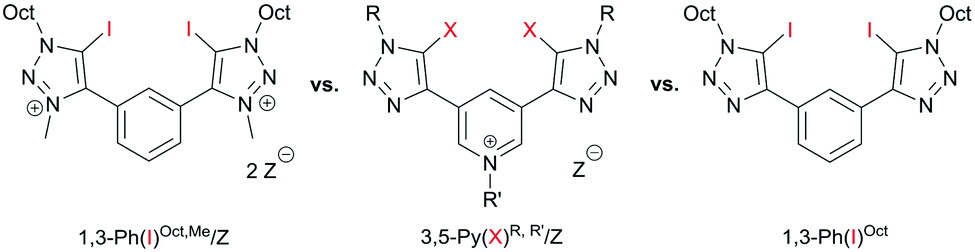 | ||
| Fig. 1 Previously used dicationic halogen bonding based activator (left), monocationic (middle) and neutral variant of this study (right). Z = triflate or BArF4. | ||
The syntheses of all XB donors employed in this study are shown in Scheme 1. First, a modified Cu(I)-catalysed azide–alkyne 1,3-dipolar cycloaddition reaction of bis-alkyne 4 with benzyl azide or octyl azide and sodium iodide led to the corresponding bis(iodotriazolyl)pyridine derivatives 3,5-Py(I)R in good yields (Scheme 1; for nomenclature see Fig. 1).15 Subsequent regioselective alkylation at the pyridine core with methyl triflate (MeOTf), octyl triflate (OctOTf) or N-methyl bis(trifluoromethyl-sulfonyl)amide (MeNTf2) provided XB donors 3,5-Py(I)R,R′/Z (Z = OTf, NTf2) in good yields (71–98%).16 In order to include a variant with noncoordinating tetrakis(3,5-bis(trifluoromethyl)phenyl)borate (BArF4), the corresponding salt 3,5-Py(I)Oct,Me/BArF4 was obtained from salt metathesis of the triflate salt with NMe4 BArF4 in chloroform in quantitative yield.16 Non-iodinated reference compounds were obtained via similar procedures (see the ESI†).
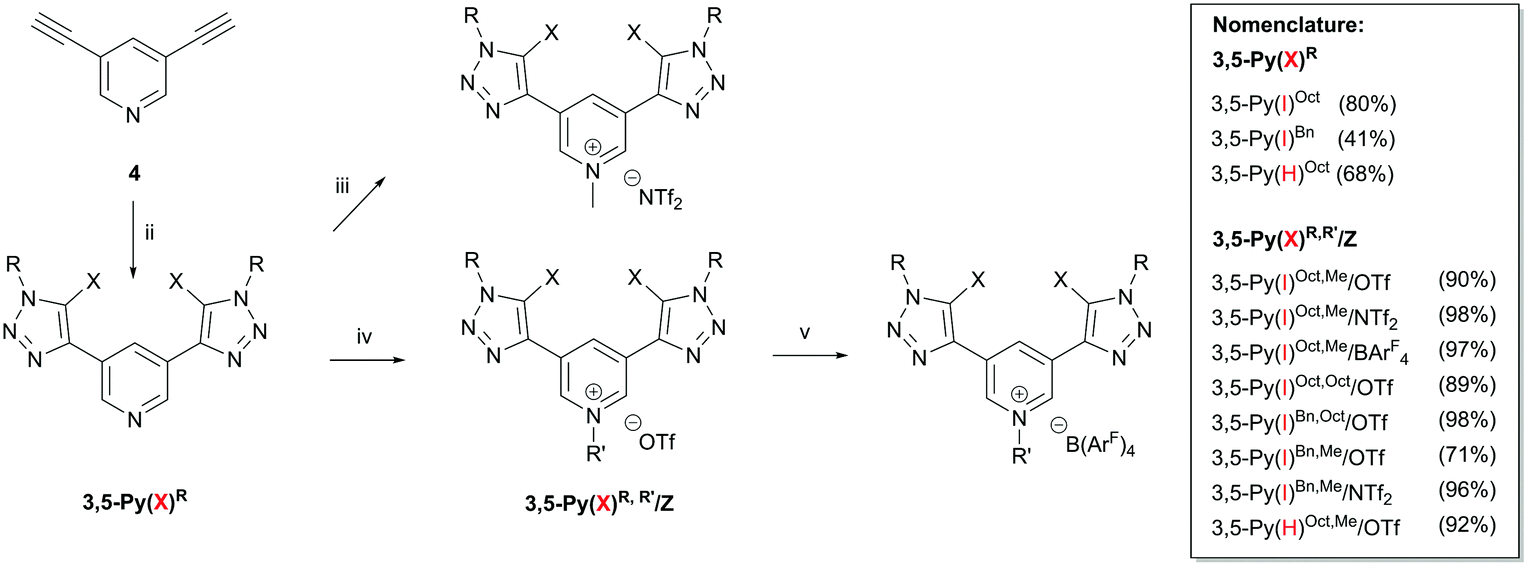 | ||
Scheme 1 Synthesis of XB donors: for X = I (ii) octyl azide or benzyl azide (2.2 eq.), CuOTf2 (4 eq.) or Cu(ClO4)2(H2O)6 (4 eq.), NaI (8 eq.), TBTA (2 mol%), DBU (2 eq.), MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF (1:1), r.t., 1–3 d. (iii) MeNTf2 (1.5 eq.), toluene:EtOAc (1:1), microwave, 130 °C, 1 h. (iv) MeOTf or OctOTf (1.2–1.5 eq.), CH2Cl2, −78 °C, 1–3 d. (v) 1.3 eq. TMA–BArF4, CHCl3, 12 h. For X = H: (ii) octyl azide or benzyl azide (2 eq.), CuSO4(H2O)5 (0.05 eq.), Na ascorbate (0.15 eq.), CH2Cl2:H2O (1:1), 5 d. (iv) MeOTf or OctOTf (1.2–1.5 eq.), CH2Cl2, −78 °C, 1 d. :THF (1:1), r.t., 1–3 d. (iii) MeNTf2 (1.5 eq.), toluene:EtOAc (1:1), microwave, 130 °C, 1 h. (iv) MeOTf or OctOTf (1.2–1.5 eq.), CH2Cl2, −78 °C, 1–3 d. (v) 1.3 eq. TMA–BArF4, CHCl3, 12 h. For X = H: (ii) octyl azide or benzyl azide (2 eq.), CuSO4(H2O)5 (0.05 eq.), Na ascorbate (0.15 eq.), CH2Cl2:H2O (1:1), 5 d. (iv) MeOTf or OctOTf (1.2–1.5 eq.), CH2Cl2, −78 °C, 1 d. | ||
The activity of these XB donors was then tested in the benchmark reaction shown in Scheme 2.17 Similarly to our previous studies, the XB donor activates the C–Br bond of benzhydryl bromide 1 to generate the benzhydryl cation. Deviating from our earlier benchmark reaction, which featured the solvolysis of the intermediate, in this study the cation was quenched with one equivalent of 1,3,5-trimethoxybenzene 2 as an external nucleophile. A stoichiometric amount of the respective XB donor was used and Cs2CO3 was added to each reaction to rule out hidden acid catalysis.
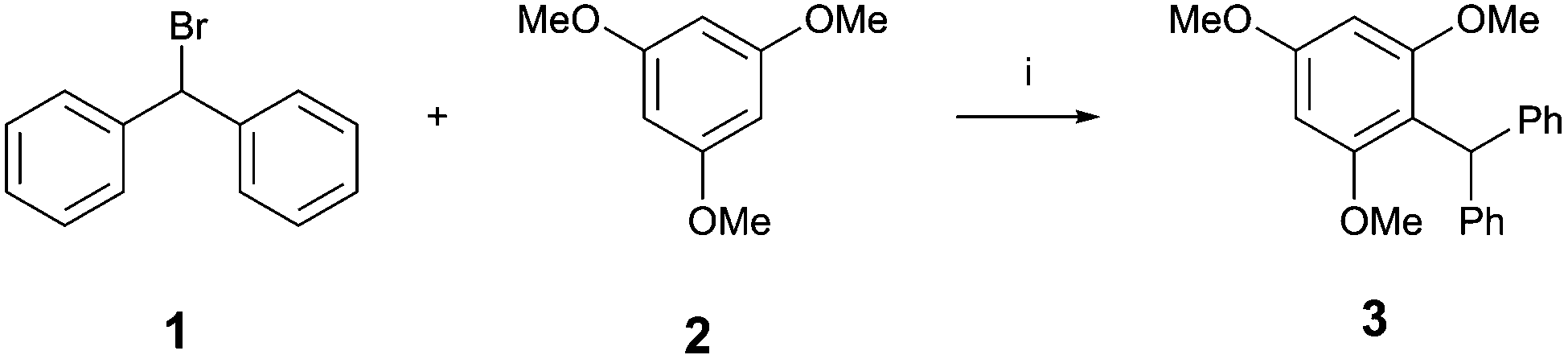 | ||
| Scheme 2 Friedel–Crafts alkylation as the benchmark reaction for the activation of the carbon–bromine bond by charge-assisted halogen bond donors. (i) 1 eq. activator (XB donor or reference compound), 1 eq. Cs2CO3, CDCl3 (1.6 mM), rt. | ||
The reaction was monitored by 1H-NMR spectroscopy (see Fig. 2 for kinetic profiles and Table 1 for yields of product 3). No significant background reaction occurs in the absence of activators (<5% yield after 30 h). Activation by Brønsted acids can also be ruled out, since 5 mol% of triflic acid (HOTf) showed no activation. As a non-iodinated reference compound 3,5-Py(H)Oct,Me/OTf induced no product formation, any activity of iodinated 3,5-Py(I)R,R′/Z compounds cannot be explained by hydrogen bonding or by simple electrostatic interactions and is thus likely based on halogen bonding.
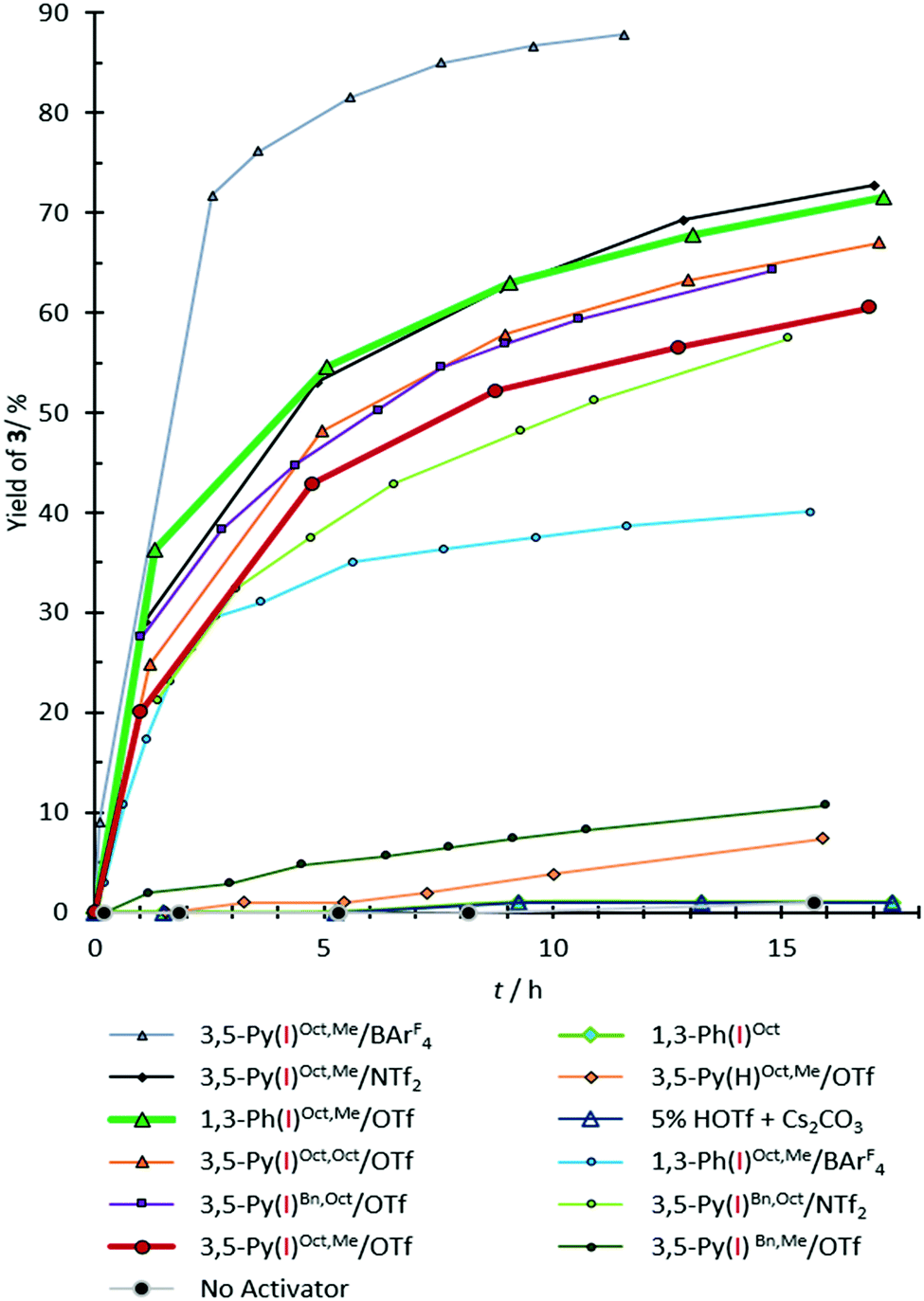 | ||
| Fig. 2 Yield vs. time diagram for the formation of compound 3 in the benchmark reaction. The yield of compound 3 according to 1H-NMR analysis. The error is approximately 5%. | ||
| # | Activating reagent | Yield [%]a (5 h)b | Yield [%]a (12 h)b |
|---|---|---|---|
| a Yields according to 1H-NMR analysis. b Partial decomposition of the XB donor. c Low solubility in CDCl3. | |||
| 1 | — | <5 | <5 |
| 2 | HOTf | <5 | <5 |
| 3 | 3,5-Py(H)Oct,Me/OTf | <5 | <5 |
| 4 | 1,3-Ph(I)Oct | <5 | <5 |
| 5 | 3,5-Py(I)Oct,Me/BArF4 | 81b | 87b |
| 6 | 3,5-Py(I)Oct,Me/OTf | 42 | 56 |
| 7 | 3,5-Py(I)Oct,Me/NTf2 | 53 | 69 |
| 8 | 3,5-Py(I)Oct,Oct/OTf | 48 | 63 |
| 9 | 3,5-Py(I)Bn,Oct/OTf | 44 | 61 |
| 10 | 3,5-Py(I)Bn,Me/NTf2 | 37 | 52 |
| 11 | 3,5-Py(I)Bn,Me/OTf | <5c | 10c |
| 12 | 1,3-Ph(I)Oct,Me/OTf | 54 | 68 |
| 13 | 1,3-Ph(I)Oct,Me/BArF4 | 35c | 38c |
The non-cationic bis(triazole) derivative 1,3-Ph(I)Oct (cf.Fig. 1) showed no activity, indicating that some form of charge assistance is essential for this reaction. The highest rate acceleration was obtained in the presence of 3,5-Py(I)Oct,Me/BArF4, yielding 72% of compound 3 after 2.5 h (Table 1, entry 5). However, after 3 h decomposition of the XB donor was observed (reaching ∼13% of the decomposed material after 11 h), and thus the interpretation of the observed performance needs some caution. In contrast, no indications of decomposition were obtained for the analogous triflate and NTf2− salts, but – as could be expected from the more coordinating nature of the counterions – these activators also induced somewhat less product formation (42% for 3,5-Py(I)Oct,Me/OTf after 5 h and 53% for 3,5-Py(I)Oct,Me/NTf2 after the same time; see Table 1, entries 6 and 7). Variation of the alkylation pattern did not decisively alter the overall outcome: changing the methyl group on the pyridinium to octyl led to a slight increase in yield, which is likely still within the error of experiment (48% yield for 3,5-Py(I)Oct,Oct/OTf after 5 h, entry 8). Substituting the octyl groups on the triazolium moieties with benzyl groups again led to virtually identical results for the NPy–Oct compound 3,5-Py(I)Bn,Oct/OTf (44% yield, entry 9). The corresponding benzylated NPy–Me compounds showed slightly (37% yield for 3,5-Py(I)Bn,Me/NTf2) to drastically lower performance (no yield for 3,5-Py(I)Bn,Me/OTf). At least in the latter case, this can be attributed to the low solubility of the XB donor in chloroform.
Most importantly, the previously studied6 dicationic XB donor 1,3-Ph(I)Oct,Me/OTf was only slightly more active (54% yield after 5 h) than its monocationic analogue 3,5-Py(I)Oct,Me/OTf (42%, see above).18 This clearly demonstrates that bidentate charge-assisted halogen bonding does not automatically also necessitate dicationic XB donors. A comparable performance as an activator is also feasible for monocationic species. In the current example, this is particularly noteworthy since the Lewis structure formalism (!) does not indicate the delocalization of the cationic charge towards the triazole moieties.
The results of the activation studies are supported by isothermal titration calorimetry (ITC) measurements (see Table 2). Both the monocationic XB donor 3,5-Py(I)Oct,Me/OTf and its dicationic variant 1,3-Ph(I)Oct,Me/OTf feature nearly the same binding strength to bromide in chloroform (1.6 × 105 M−1, entries 2 and 10). The binding to chloride is also very similar (entries 1 and 9), whereas a notable deviation is found for iodide complexation. Somewhat surprisingly, the monocationic system 3,5-Py(I)Oct,Me/OTf turned out to be the stronger Lewis acid. A clear difference between the two types of XB donors is the relative contribution of the enthalpic and entropic parts. For the monocationic XB donor, the enthalpic part is decidedly more favourable compared to 1,3-Ph(I)Oct,Me/OTf. On the other hand, the entropic contribution is negligible for the former, but constitutes an important favourable contribution in the latter (similar to the binding of bis(iodoimidazolium)-derived XB donors).19
| # | Host | Xa | K [M−1] | ΔH [kJ mol−1] | −TΔS [kJ mol−1] |
|---|---|---|---|---|---|
| a Tetraoctylammonium salts were used as guests; c(guest) = 10 mM; c(host) = 1 mM. The guest solution was titrated into the host solution. b No accurate fitting data could be obtained. | |||||
| 1 | 3,5-Py(I)Oct,Me/OTf | Cl | 1.35 × 105 | −19.34 | −10.42 |
| 2 | 3,5-Py(I)Oct,Me/OTf | Br | 1.59 × 105 | −21.50 | −8.67 |
| 3 | 3,5-Py(I)Oct,Me/OTf | I | 9.26 × 104 | −21.77 | −7.05 |
| 4 | 3,5-Py(I)Oct,Me/NTf2 | Cl | 2.53 × 105 | −25.41 | −5.94 |
| 5 | 3,5-Py(I)Oct,Me/NTf2 | Br | 2.59 × 105 | −24.66 | −6.74 |
| 6 | 3,5-Py(I)Oct,Me/NTf2 | I | 1.53 × 105 | −25.66 | −4.42 |
| 7 | 3,5-Py(I)Oct,Me/BArF4 | Cl | 3.16 × 105 | −24.25 | −7.67 |
| 8 | 3,5-Py(I)Oct,Me/BArF4 | Br | 3.57 × 105 | −29.15 | −3.06 |
| 9 | 1,3-Ph(I)Oct,Me/OTf | Cl | 9.09 × 104 | −12.45 | −15.85 |
| 10 | 1,3-Ph(I)Oct,Me/OTf | Br | 1.60 × 105 | −14.36 | −15.81 |
| 11 | 1,3-Ph(I)Oct,Me/OTf | I | 1.82 × 104 | −17.68 | −12.81 |
| 12 | 1,3-Ph(I)Oct,Me/BArF4 | Cl | —b | — | — |
| 13 | 1,3-Ph(I)Oct,Me/BArF4 | Br | —b | — | — |
As had already been observed,13 the association strength decreases in the order chloride > bromide > iodide for all XB donors tested. The influence of the counterion could only be investigated for the monocationic species, as the dicationic ones suffered from solubility issues. As expected, the binding strength of the XB donors follows the lesser Lewis basicity of the counterions (BArF4− > NTf2− > OTf−, entries 1–8). This underlines the relevance of ion pairing and may also explain the similar Lewis acidity (and activation potential) of mono- and dicationic XB donors: ion pairing in the latter is likely stronger, which might compensate for their potentially stronger XB donor properties.
Finally, X-ray structural analyses helped to shed light on the binding mode of the monocationic XB donors to anions. The result of the single-crystal X-ray analysis of the XB donor 3,5-Py(I)Bn,Me/OTf is shown in Fig. 3. Two strong halogen bonds between iodine atoms of the XB donor and oxygen atoms of triflate can be found in the solid state. The iodine–oxygen bond distances (2.78 Å and 2.99 Å) are shorter than the sum of the van der Waals radii (3.5 Å)20 and the binding angle C–I–O is nearly linear (173° and 166°), as expected for halogen bonding. The previously reported crystal structure of the dicationic XB donor 1,3-Ph(I)Bn,Me/OTf featured halogen bonds to triflate anions with I–O distances of 2.88 Å and 2.90 Å.8
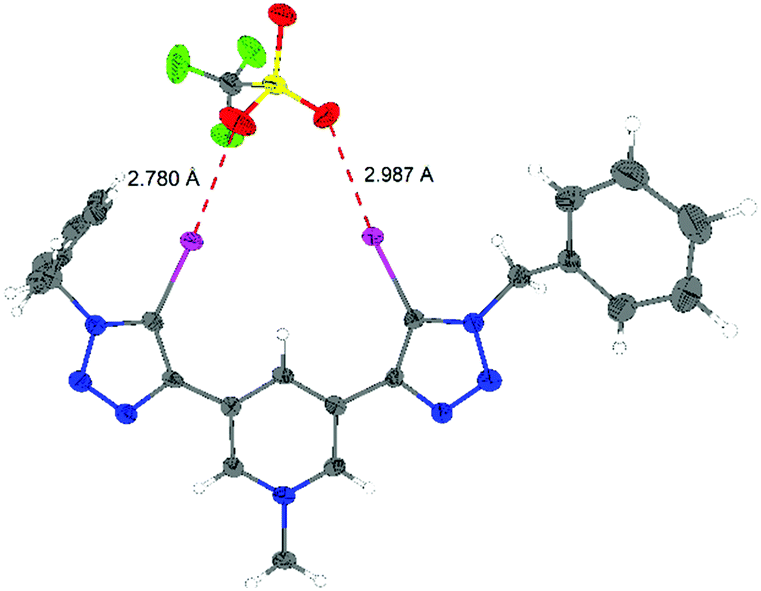 | ||
| Fig. 3 ORTEP21 plot of the X-ray structural analysis of compound 3,5-Py(I)Bn,Me/OTf (ellipsoids at 50% probability). Selected bond distances [Å] and angles [°]: I1–O3 = 2.780, I2–O2 = 2.987, I1–C8 = 2.056, I2–C17 = 2,072, C8–I1–O3 = 173.62, and C17–I2–O2 = 166.57. | ||
From a solution of 3,5-Py(I)Bn,Oct/OTf and benzhydryl bromide (1) in acetonitrile, we obtained a single-crystal of 3,5-Py(I)Bn,Oct/Br (Fig. 4). The bromide is bound in a bidentate fashion by the XB donor with I–Br distances of 3.19 Å and 3.23 Å. These distances are again significantly shorter than the sum of the van der Waals radii (3.83 Å)20 and the binding angle C–I–Br is close to linear (171° and 166°). A similar crystal structure of 3,5-Py(I)Oct,Me/Cl was previously reported by Beer et al.13 In all reported crystal structures of monocationic XB donors with the 3,5-Py(I)R,R′/Z core motif, both triazolyl moieties are in plane with the pyridinium ring. In the crystal structure of 1,3-Ph(I)Bn,Me/OTf, on the other hand, the triazolium groups are out of plane with the central benzene ring.6 As a consequence of the planar geometry of the core structure of 3,5-Py(I)R,R′, π conjugation will allow some delocalization of the cationic charge.
 | ||
| Fig. 4 ORTEP21 plot of the X-ray structural analysis of compound 3,5-Py(I)Bn,Oct/Br (ellipsoids at 50% probability). Selected bond distances [Å] and angles [°]: I1–Br = 3.185, I2–Br = 3,227, I1–C22 = 2.075, I2–C32 = 2,091, C22–I1–Br = 171.70, and C32–I2–Br = 166.51. | ||
In summary, a comparison of halogen bond donors based on either a bis(iodotriazolium)benzene or a bis(iodotriazolyl)pyridinium motif allowed elucidation of the influence of charge (dicationic vs. monocationic) on the Lewis acidity of these compounds. In a halide abstraction benchmark reaction, the performance of both types of XB donors was very similar. Calorimetric measurements also confirmed that overall the Lewis acidity of the monocationic XB donors is roughly in the same range as the one of the previously studied dicationic bidentate variants. The titrations also revealed that XB donors with less Lewis basic anions bind stronger to halides (BArF4− > NTf2− > OTf−), in agreement with the performance in the test reaction. X-ray structural analyses indicate that the cationic charge is likely delocalized over the planar bis(iodotriazolyl)pyridinium motif.
These findings may have important implications for the design of strong halogen-bonding-based activators or organocatalysts: apparently, a monocationic backbone is sufficient to ensure strong charge-assisted bidentate halogen bonding. The monocationic species, however, feature some distinct advantages over their respective dicationic analogues: complete anion exchange by metathesis is realized more easily, and the solubility in less polar organic solvents is generally superior. This already became apparent in this study, as 3,5-Py(I)Oct,Me/BArF4 exhibits much better solubility in chloroform than 1,3-Ph(I)Oct,Me/BArF4 (see footnote 18). As a further example, 3,5-Py(I)Oct,Oct/OTf exhibits much better solubility in diethylether than 1,3-Ph(I)Oct,Me/OTf. As a consequence, the utilization of monocationic motifs like the one studied here might allow to combine the “best of both worlds” in halogen-bonding organocatalysis: the performance of charge-assisted XB donors and the solubility of (generally weaker) fluorinated XB donors in less polar media.
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (Grant Agreement No. 638337) and from the Fonds der Chemischen Industrie.
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711 CrossRef CAS.
- D. Bulfield and S. M. Huber, Chem. – Eur. J., 2016, 22, 14434 CrossRef CAS PubMed.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118 CrossRef CAS PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478 CrossRef CAS PubMed.
- N. L. Kilah, M. D. Wise, C. J. Serpell, A. L. Thompson, N. G. White, K. E. Christensen and P. D. Beer, J. Am. Chem. Soc., 2010, 132, 11893 CrossRef CAS PubMed.
- F. Kniep, L. Rout, S. M. Walter, H. K. V. Bensch, S. H. Jungbauer, E. Herdtweck and S. M. Huber, Chem. Commun., 2012, 48, 9299 RSC.
- S. M. Walter, F. Kniep, E. Herdtweck and S. M. Huber, Angew. Chem., Int. Ed., 2011, 50, 7187 CrossRef CAS PubMed.
- F. Kniep, S. M. Walter, E. Herdtweck and S. M. Huber, Chem. – Eur. J., 2012, 18, 1306 CrossRef CAS PubMed.
- R. Tepper, B. Schulze, P. Bellstedt, J. Heidler, H. Gorls, M. Jager and U. S. Schubert, Chem. Commun., 2017, 53, 2260 RSC.
- R. Tepper, B. Schulze, M. Jäger, C. Friebe, D. H. Scharf, H. Görls and U. S. Schubert, J. Org. Chem., 2015, 80, 3139 CrossRef CAS PubMed.
- S. M. Walter, S. H. Jungbauer, F. Kniep, S. Schindler, E. Herdtweck and S. M. Huber, J. Fluorine Chem., 2013, 150, 14 CrossRef CAS.
- S. H. Jungbauer, S. M. Walter, S. Schindler, L. Rout, F. Kniep and S. M. Huber, Chem. Commun., 2014, 50, 6281 RSC.
- S. W. Robinson, C. L. Mustoe, N. G. White, A. Brown, A. L. Thompson, P. Kennepohl and P. D. Beer, J. Am. Chem. Soc., 2015, 137, 499 CrossRef CAS PubMed.
- Triply-charged systems featuring two iodotriazolium groups next to the pyridinium core were also prepared, but suffered from very bad solubility and decompositon (dealkylation).
- W. S. Brotherton, R. J. Clark and L. Zhu, J. Org. Chem., 2012, 77, 6443 CrossRef CAS PubMed.
- S. H. Jungbauer and S. M. Huber, J. Am. Chem. Soc., 2015, 137, 12110 CrossRef CAS PubMed.
- N. Watanabe, A. Matsugi, K. Nakano, Y. Ichikawa and H. Kotsuki, Synlett, 2014, 438 CAS.
- For the corresponding BArF4 compounds, even a slight decrease in activity is observed for the dicationic XB donor 1,3-Ph(I)Oct,Me/BArF4 (35% yield after 5 h), again likely due to low solubility.
- S. M. Walter, F. Kniep, L. Rout, F. P. Schmidtchen, E. Herdtweck and S. M. Huber, J. Am. Chem. Soc., 2012, 134, 8507 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Syntheses as well as kinetic, titration and crystallographic data. CCDC 1817655 and 1817656. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc00527c |
| This journal is © The Royal Society of Chemistry 2018 |